PDF File: (Click to Down Load): Chapter6.pdf
Related Web Pages you should Look at:
British Site
German Site
Physics Approach
A Nicely Organized Page
Physics Details
Introduction:
Nuclear magnetic resonance is an absorption spectroscopy involving the absorption of radio frequency EM waves. Since we have already covered IR absorption spectroscopy it is appropriate to compare these two techniques, building on what we already know. The energy associated with a photon in the radio frequencies is extremely small compared to IR frequencies. In fact, we are constantly being irradiated by radiowaves with no effect. Absorption spectroscopies rely on the transfer of energy (h[nu]) from an electro-magnetic wave to a quantized transition in a material. For IR the quantized transition (a transition with a fixed energy) is the vibration of a chemical bond.
NMR involves changes in the spin state of the nucleus of an atom. Not all nuclei display spin. In order to display spin a nucleus must have an odd number of protons or neutrons. Hydrogen has one proton, so displays spin (1H or proton NMR). Deuterium has one proton and one neutron and also displays spin. Other atoms that display spin are isotopes of common elements (deuterium is an isotope of hydrogen). The most common are 13C, 19F, 15N and 29Si. An atom with spin has a non-zero spin quantum number, I. For most nuclei of interest to polymer scientists the spin quantum number is 1/2. Deuterium and Nitrogen 15 have spin quantum numbers of 1. The number of spin states possible for a nucleus is given by 2I+1. For most nuclei of interest to polymer scientists there are two spin states. IR absorption also involves two states, i.e. vibrating and not vibrating. In NMR the two states correspond with two orientations of magnetic moment vector for the nucleus with respect to an external magnetic field as discussed below.
For a quantized transition to occur a system must be constrained, i.e. a guitar string must be under tension for the production of a note, atoms must be bonded for an IR absorption. In NMR the constraint which leads to quantized transitions is applied to the sample externally in a large static magnetic field. If a nucleus spins and is composed of charged particles it possesses a magnetic moment associated with the angular velocity of the charged particles. The magnitude of the magnetic moment, u, is proportional to the spin quantum number,
![]()
gN is a constant and [beta]N is the nuclear magneton, given by,
![]()
where all the parameters are constant and related to a proton. The magnetic moment is a vector, u, and in vector form it is defined as,
![]()
where I is the angular momentum vector, given by.
![]()
and [gamma] is the magnetogyric ratio for the nucleus. The frequency of absorption in IR is directly proportional to the magnetiogyric ratio,
![]()
where B0 is a strong magnetic field which is applied to the sample.
The nucleus can then be thought of as a magnet, and in the absence of a magnetic field these tiny magnets are randomly arranged in a sample with no preferred direction for the magnetic moment vectors. Application of a radiofrequency EM wave to such a sample has no effect, i.e. there is no absorption. There is not absorption because the system can no tell the difference between magnetic vectors pointing up or down or any other direction since these directions have not reference base. This is analogous to a guitar string which is not constrained or two atoms which are not bonded in IR. In the absence of constraints there is no perceptible absorption.
If a strong magnetic field is applied to the nuclei, they can tell the difference between alignment in the direction of the applied magnetic field and opposed to the applied magnetic field. In NMR the constraint which leads to quantized transitions is applied by the spectrometer. Because of this the frequency of absorption varies with the applied magnetic field and there is no absolute frequency or wavelength for a given absorption. NMR spectra are not plotted as absorption versus wavenumber as IR spectra are, they are plotted as absorption versus chemical shift, d. The chemical shift for proton NMR is the difference between the frequency of absorption of the sample and a standard, tetramethylsilane (TMS) normalized by the frequency of absorption of TMS,
![]()
d is expressed in parts per million (ppm) so the above equation is multiplied by 106.
In IR we consider two states for a bond, vibrating and non-vibrating. The transition associated with the change from non-vibrating to vibrating leads to the absorption at fixed wavenumbers. In NMR several states are potentially possible depending on the spin quantum number. The permitted states are given by the allowable values for the magnetic quantum number, mI = I, I-1,...-I. For I = 1/2 there are two states possible, mI = 1/2 and mI = -1/2. For I = 1 three states are possible, mI = 1, 0, -1. For protons the two allowable states are generally spoken of as parallel and anti-parallel to the applied field.
Intensity of Absorption:
The strength of an IR absorption band depends on the change in dipole moment for the bond on vibration, i.e. how polar a bond is. The strength of a NMR absorption band depends on the magnitude of the magnetogyric ration, [gamma], i.e. how large the magnetic dipole moment is. The absorption in IR is also proportional to the concentration of the absorbing bond. NMR depends on the presence of specific isotopes. In considering the strength of a NMR absorption band we consider the "natural abundance" of these isotopes. For example, 99.98 percent of hydrogen atoms are 1H, and 0.0156 percent are deuterium, 2H. The magnetogyric ratio for hydrogen is 26,700 while for deuterium is 4,100. This means that proton NMR (1H) results in 100 times the signal as deuterium if a sample contains natural abundance hydrogen. A similar comparison shows that proton NMR absorption is about 50 times stronger than 13C NMR (natural abundance of 13C is about 1%).
An NMR absorption peak for a given nucleus is directly proportional to the number of these atoms in a sample.
A major difference between analysis of NMR spectroscopy in polymers and IR spectra is that all absorption bands in NMR are uniquely identifiable.
The NMR Experiment, Pulsed NMR:
The simplest NMR experiment is the observation of "free induction decay" from a radio frequency pulse. This is analogous to the plucking of a guitar string. The free induction decay looks similar to the raw data obtained form a FTIR instrument except that the x-axis is time rather than space. Fourier transform of the spectra results in inverse units, i.e. frequency, which is converted to d.
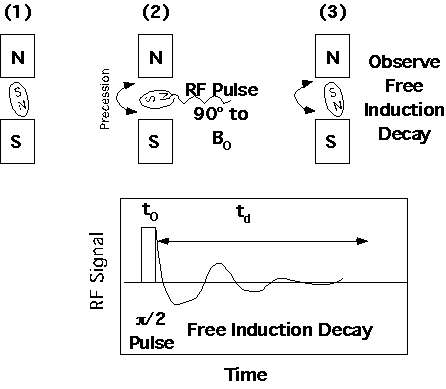
The NMR instrument is capable of many other more complicated experiments involving sequencing of pulses and observation of kinetic phenomena in spin relaxation which will be discussed later in this chapter.
The nuclear dipole tilts at an angle [theta] with respect to the static magnetic field when the RF pulse is applied. The magnetic dipole then rotates about the static field at this precession angle, [theta]. The precession angle is determined by the RF field strength, H1, the pulse time, t0, and the magnetogyric ration of the nucleus, [gamma].
![]()
Position of Absorption Peaks in NMR Spectra:
Consider an isolated nucleus in a static magnetic field of B0 = 14,000 Gauss. The frequency of absorption for different nuclei varies according to the magnetogyric ratio,
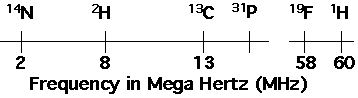
NMR is extremely sensitive to the "chemical environment" of a nucleus. "Chemical environment" means the local magnetic environment. The local magnetic environment is changed by "shielding" or "deshielding" depending on how the chemical bonds which are attached to the nucleus withdraw electrons from the electron cloud of a bare atom. Electron withdrawing groups such as the aromatic ring, deshield a proton for instance and give rise to a deshielded proton with a large d. Tetramethyl silane (TMS) has highly shielded protons so is used as a standard for the 0 point of a NMR spectra. The only absolute point for a given nucleus would be a nucleus stripped of all electrons. Since it is not possible to obtain such a completely deshielded nucleus TMS is used as a standard.
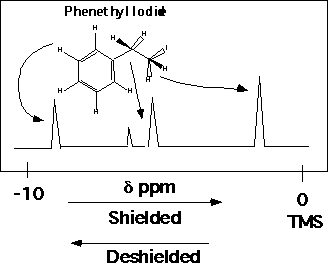
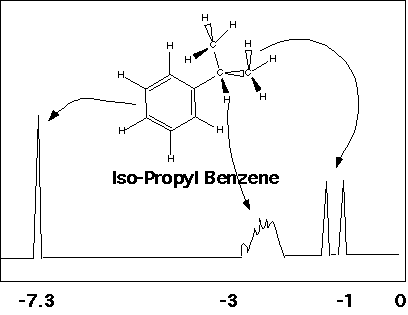
The "Chemical Environment" reflects the bonding geometry of a molecule. Two protons with identical chemical environments "see" the same view of the molecule from their position in the bonding structure. The same view includes the stereochemical arrangement of the molecule, i.e. the handedness of the structure. The "view" which effects proton absorption is three bonds in distance, that is, structural differences more than three bonds away don't matter. Additionally, the presence of resonance structures such as the aromatic ring in phenethyl iodide makes all of the aromatic protons see the same chemical environment.
The aromatic protons in phenethyl iodide are identical. The methyl iodide group at the other end of phenethyl iodide has two protons with identical chemical environments, so give rise to a single peak at the far right of the spectrum. The two methylene protons have different chemical environments because of their stereochemical relationship to the methyl iodide group.
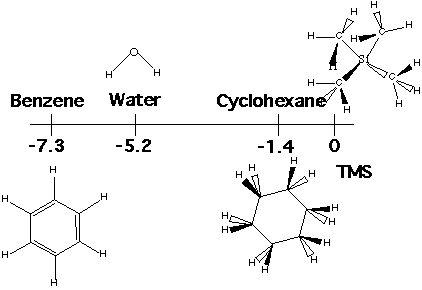
Protons with the same chemical environment give rise to a single peak, so simple or symmetric molecules have single peaks in proton NMR spectra. Examples are water, benzene and cyclohexane. The aromatic ring in benzene is strongly electron withdrawing, so the protons are strongly deshielded. The oxygen in water is also electron withdrawing and deshielding. The cyclohexane ring is only weakly electron withdrawing so weakly deshielding in proton NMR.
Definition of Magnetic Equivalence:
Protons are equivalent if:
1) Same chemical shift, d.
2) Same coupling constant to all nuclei in the molecule, JHa.
Examples:
(CH3)2CHBr 2 types CH3 and CH
Propene CH3CHCH2
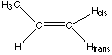 4 types Hcis, Htrans, CH3, CH
4 types Hcis, Htrans, CH3, CH
Magnetic Equivalence Means Same Magnetic Environment
There is not way to magnetically distinguish the nuclei
The Nuclei's view of the molecule is the same both in terms of bonds and in terms of stereochemistry. Nuclei can only sense 3 bonds away, with no conjugated double or triple bonds between them and can only sense other nuclei with similar nuclear magnetic transitions.
n Relative Intensity of split bands.
1 1 1
2 1 2 1
3 1 3 3 1
4 1 4 6 4 1
5 1 5 10 10 5 1
6 1 6 15 20 15 6 1
For coupling by non-chemically identical protons the binomial distributions multiply, i.e. for splitting by two non-chemically identical protons 4 bands of equal intensity would result.
Unlike IR, all absorption bands in an NMR spectrum can be completely identified. This makes NMR an extremely powerful tool for chemical identification. NMR is also extremely flexible since you have complete control over the constraint which leads to the absorption. The drawback to NMR is the cost of the instrument, $250,000 to $1e6, and the expertise needed for more than routine identification. The cost and expertise are roughly both an order of magnitude higher than IR.
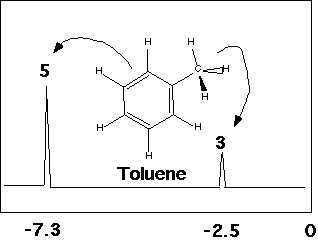
Example: Ethanol
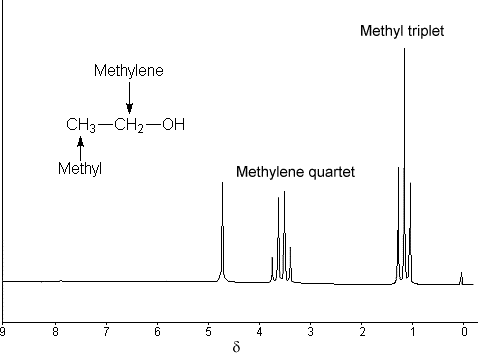
Shielding is directly related to the electron density around the nucleus. There are a number of homologous series (series varying a chemical group to observe the chemical shift) which demonstrate this.
Chemical Shifts/Electronegativity for CH3CH2X
X Electronegativity([chi]) Chemical Shift d for CH2
-SiEt3 1.9 0.6
-H 2.2 0.75
-CEt3 2.5 1.3
-NEt2 3.0 2.4
-OEt 3.5 3.3
-F 4.0 4.0
Double bonds and Rings with conjugated bonds (high electron orbitals) deshield associated protons:
Compound Structure Chemical Shift d for CH2
ethane CH3CH3 0.9
ethylene CH2=CH2 5.0 (Current)
ethyne HC=CH 2.3 (No Current)
Benzene [phi]-H 7.3 (Current)
Parts of the NMR Spectra:
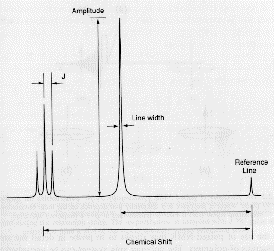
In summary the NMR spectra is composed of absorption peaks at a value of chemical shift, d, relative to a reference material, usually TMS. The amplitude is proportional to the number of magnetically equivalent protons of that type in the molecule. When neighboring protons (3 bonds or less away) are present the peak amplitude is split into several peaks following the binomial distribution and separated by the J coupling constant which is fixed in value, in terms of frequency, of the J coupling (not d) according to the type of proton which is causing the splitting (see figure below):
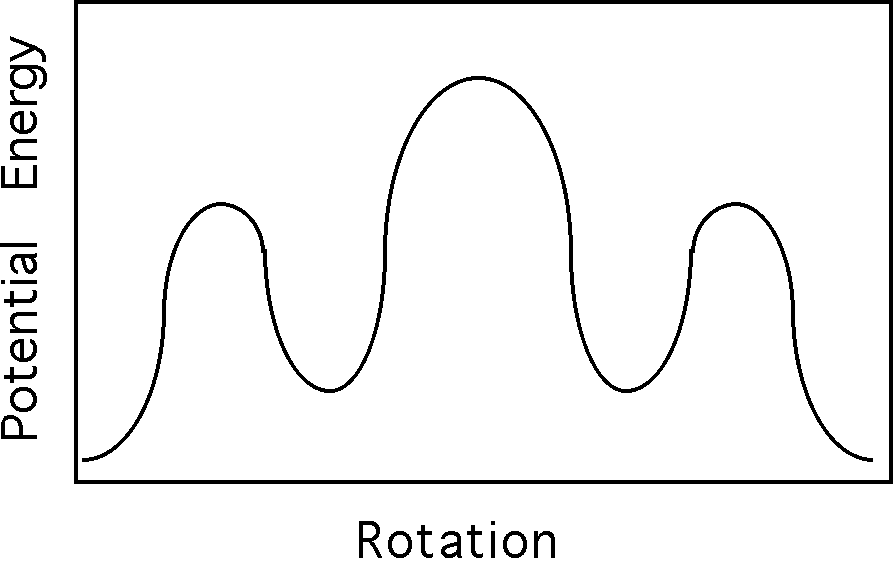
Tacticity:
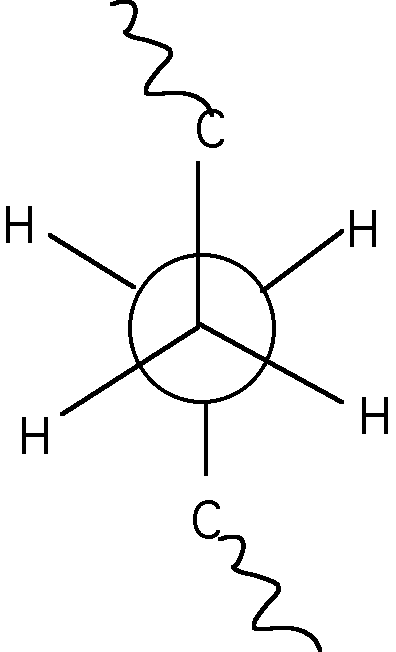
The tetrahedral bond of carbons can be thought of as a tripod with a bond sticking straight up. If a polymer chain is attached to the bond sticking straight up and the chain is attached to one of the legs of the tripod then substitutent groups attached to the other two legs have a choice of being placed to the right or left. This can be depicted in a Neuman projection along the chain back bone where the circle and three line apex are two carbons along the main chain connected by a bond.

Diad Tacticity:
For two substitutent groups in a three carbon sequence the substitutents can be located with the same handedness (Meso) or with opposite handedness (Racemic).
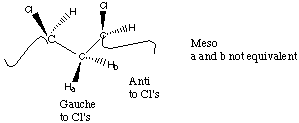
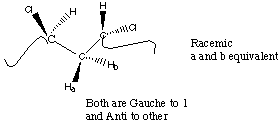
From an NMR perspective, Racemic diads give rise to two magnetically equivalent protons on the methylene group while Meso diads give rise to two magnetically different protons on the methylene group.
NMR can not sense diad tacticity because the proton on the substituted carbon can sense two diads (on either side). The smallest unit of tacticity which NMR can detect in polymers is a triad (3 mer units). Because of this triad tacticity is the usual way to refer to polymer stereochemistry. NMR can also sense higher odd number groupings of tactic mer units, with diminishing resolution, pentads (5), heptads (7) etc.
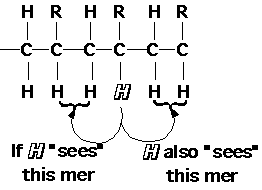
Isotactic, meso+meso mm 1
Syndiotactic, racemic+ racemic rr 1
Heterotactic, racemic + meso or meso + racemic rm or mr 2
A polymer with no preferred tacticity, an atactic polymer, has a random statistical distribution of diad tacticities so it would have 25% isotactic triads, 25% syndiotactic triads and 50% heterotactic triads. A polymer with 50% meso and 50% racemic diads does not necessarily have an atactic triad distribution, just as an atactic triad distribution does not necessarily have an atactic (random) pentad distribution. An atactic (random) pentad distribution does imply atactic triad and diad distributions.
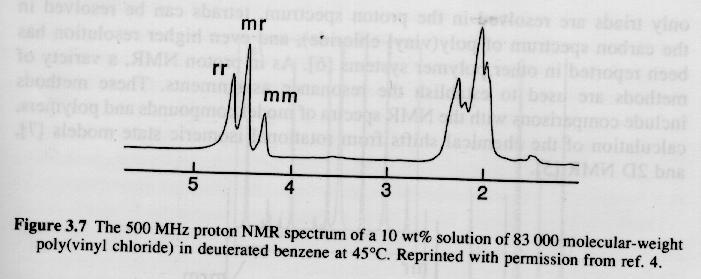
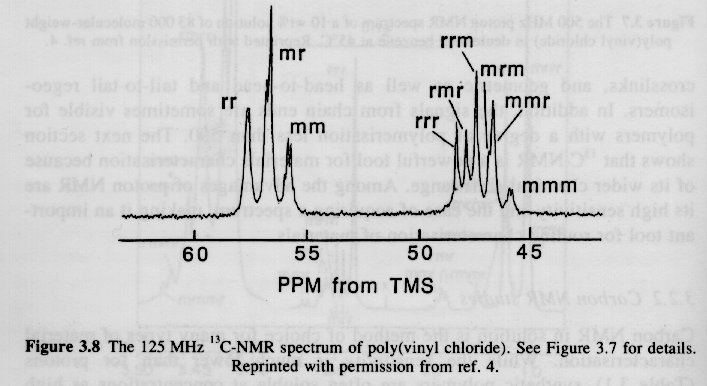
Since syndiotactic is composed of two racemic units, and because the protons in a racemic diad are magnetically equivalent (see above), then syndiotactic triads will have the fewest number of magnetic types of protons and the fewest peaks and splittings. This is shown for PMMA in figure 6.6 of Campbell and White shown below (isotactic top, syndiotactic bottom):
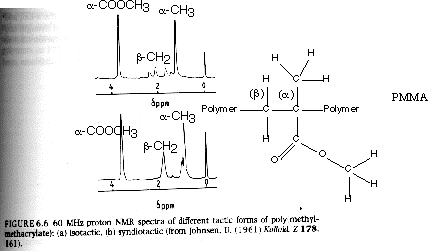
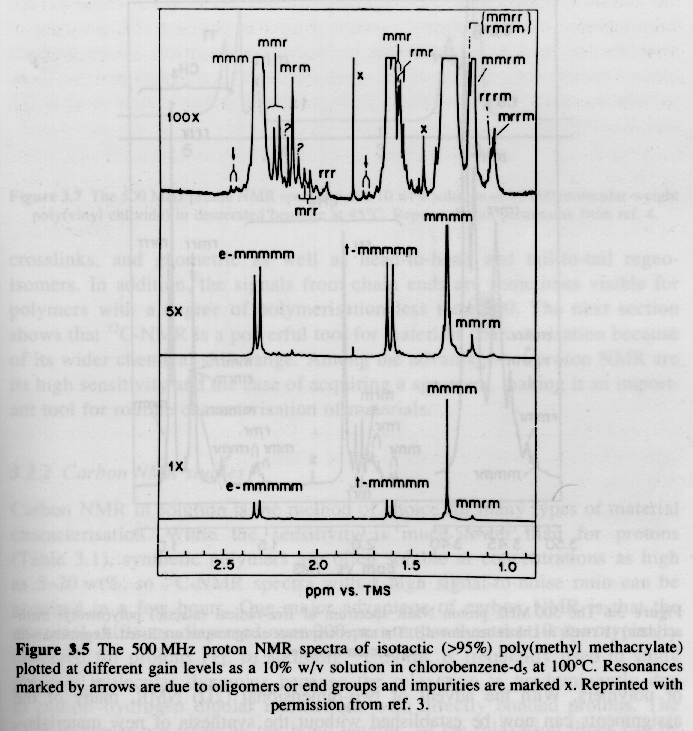
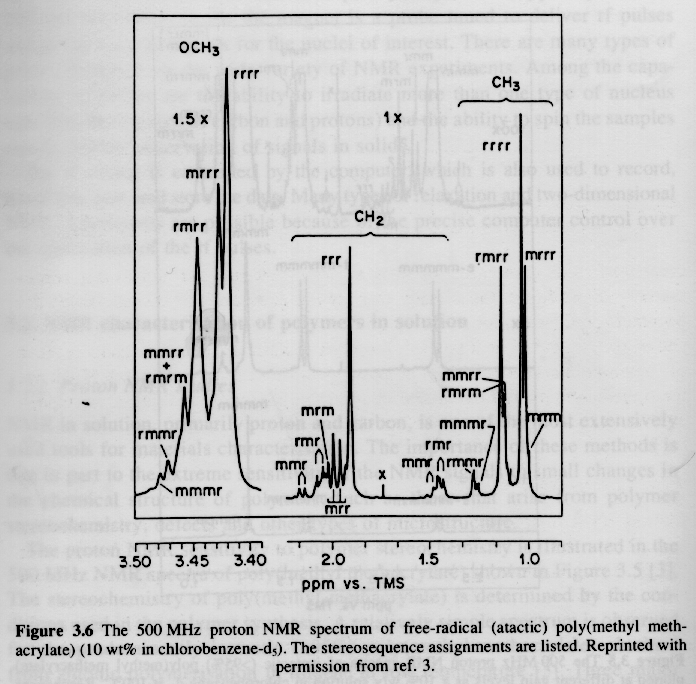
From Hunt and James "Polymer Characterization"
13C NMR:
1) Low Natural Abundance: Since most polymers are composed of hydrogen and carbon, the natural alternative nucleus for NMR is 13C. There are a number of major differences between proton and carbon 13 NMR. First, the natural abundance of 13C is much lower than 1H (12C does not display spin since the number of protons and neutrons are both even). The natural abundance of 13C is about 1.1 % while that of 1H is close to 100%. Since only nuclei of similar magnetic resonance can lead to coupling and splitting of the absorption peaks, the low natural abundance of 13C leads to no splittings of the absorption peaks. The sensitivity of absorption of a RF pulse and the associated decay are also much lower for 13C.
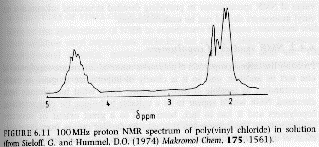
2) Large Chemical Shifts: The range of proton absorptions are on the order of 10ppm relative to TMS. For 13C the range of absorptions are on the order of 200ppm relative to TMS. The 13C spectrum has more than an order higher resolution when compared to 1H spectra as can be seen in the PVC spectra above.
3) The large abundance of 1H nuclei compared with 13C leads to loss of 13C resolution and signal due to weak coupling of 13C and 1H resonances. This problem is amplified in solid samples, so called solid state 13C NMR.
Cross Polarization:
The low abundance of 13C leads to poor absorption of the RF pulse in a FT-NMR experiment. This limitation can be over come by exciting the protons in a sample followed by a sequence of two series of long-time pulses which make the 13C and 1H nuclei resonate at the same frequency. The latter is called the "Hartman-Hahn" condition and the process is called "cross-polarization" and the time of cross polarization is called the "contact time" or "spin-lock time". This cross polarization acts as a strong pulse for the carbon 13 nuclei (see figure below).
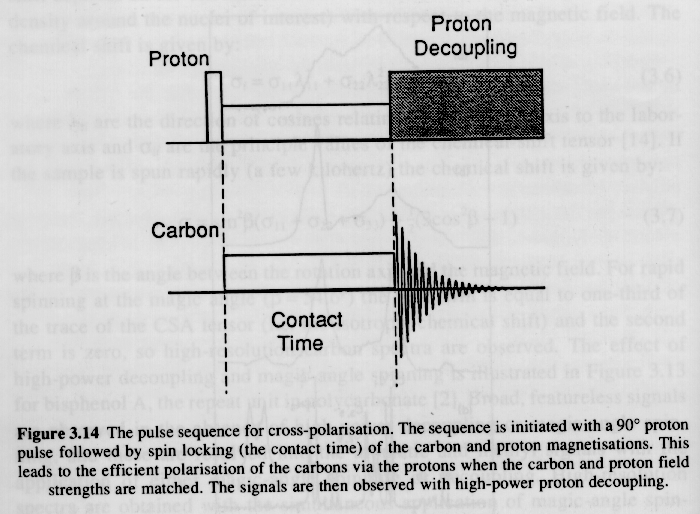
Cross-polarization leads to a large enhancement of the excitation of 13C nuclei. The large number of 1H in the sample, however, interfere with the decay of the isolated 13C nuclei due to weak interaction of the spins. For example, this would be like trying to play a guitar under water, that is despite the difference in resonance frequency for a swimming pool full of water and the guitar string, there is transfer of energy to the pool from the guitar. This dampening of the 13C signal can be removed by a strong radio frequency signal which essentially holds the protons in a highly resonating state so they are not capable of absorbing resonance from 13C nuclei. Cross polarization and spin decoupling were critical developments for the wide use of 13C NMR. The figure below shows the effect of proton decoupling on a carbon 13 NMR signal.
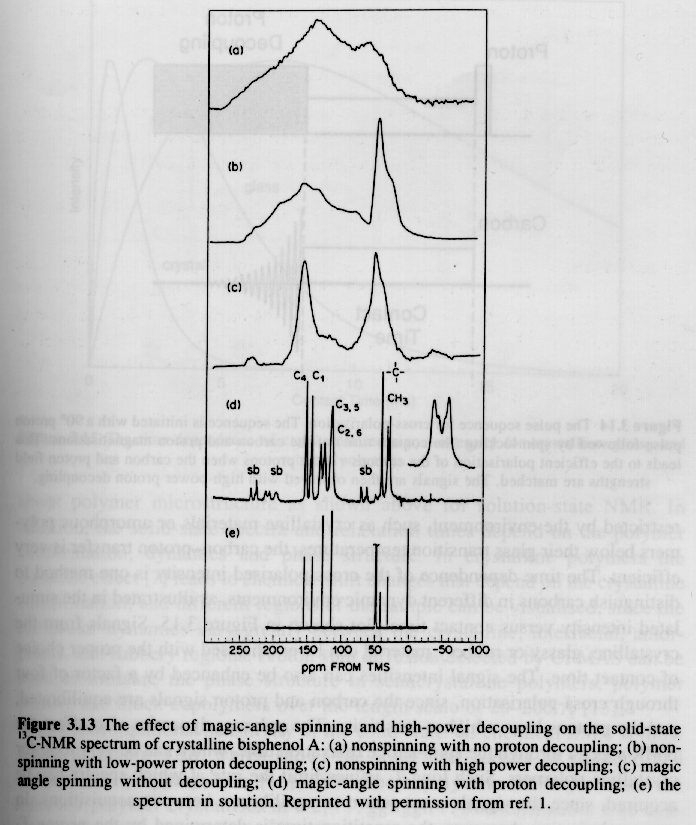
All of the previous discussion was based on "solution NMR" where a polymer sample is dissolved in a solvent at 1 to 20% concentration. One reason for studying polymers in solution is that the anisotropy of the magnetic moment with respect to the macromolecule (Chemical Shift Anisotropy, CSA) is averaged out due to thermal motion of the molecule in solution. Chemical shift anisotropy has the effect of smearing out the NMR signal as can be seen by comparison of the bottom and second to the bottom spectra in the previous figure. In the solid state, i.e. a semi-crystalline or glassy polymer, CSA has a severe effect on the spectra in broadening the absorption peaks and the effect becomes worse the higher the restriction of mobility of the chains or molecules. Through a tensoral analysis of the magnetic moments in a molecule it is possible to demonstrate that a "Magic Angle" exists with respect to the applied magnetic field at which rapid spinning of the solid sample leads to minimization of absorption line broadening due to chemical shift anisotropy.
Examples of 13C NMR spectra:
Several examples of 13C NMR spectra from Campbell and White, and Hunt and James are given below. The PMMA spectra should be compared with the proton NMR spectra given above.
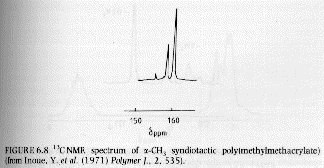
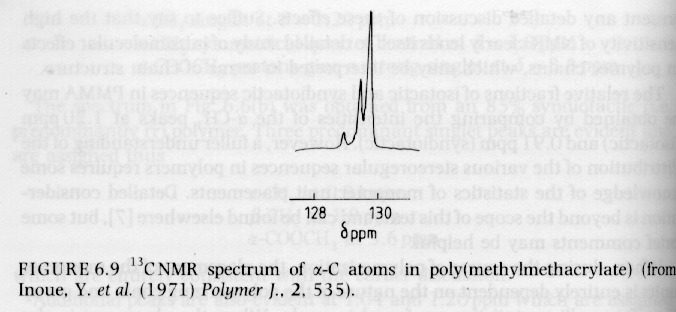
From Campbell and White "Polymer Characterization"
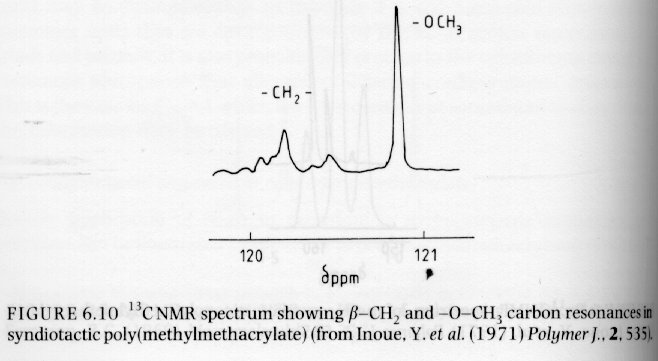
From Campbell and White "Polymer Characterization"
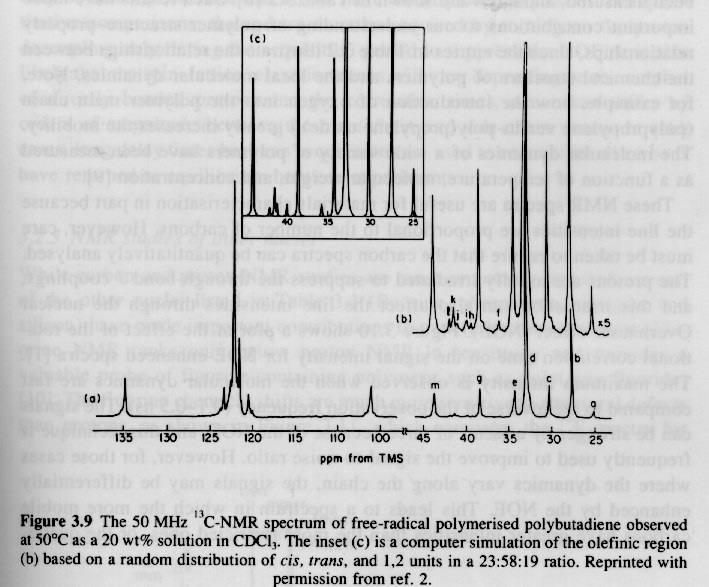
From Hunt and James "Polymer Characterization"
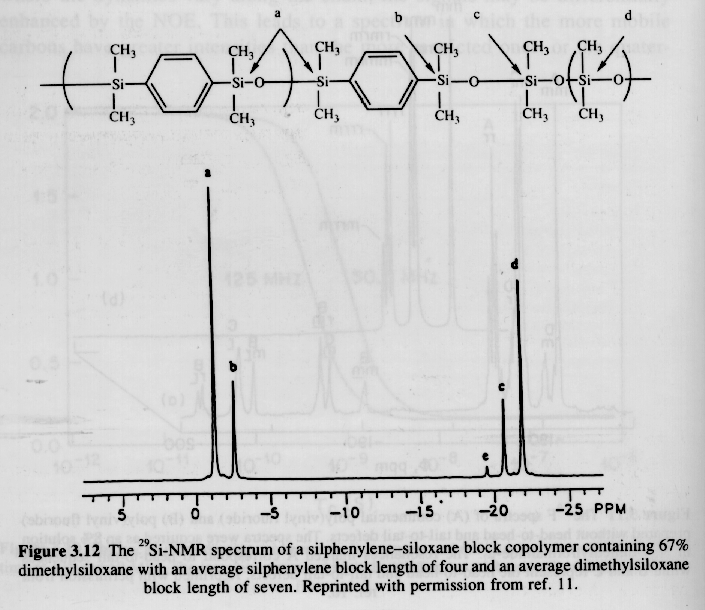
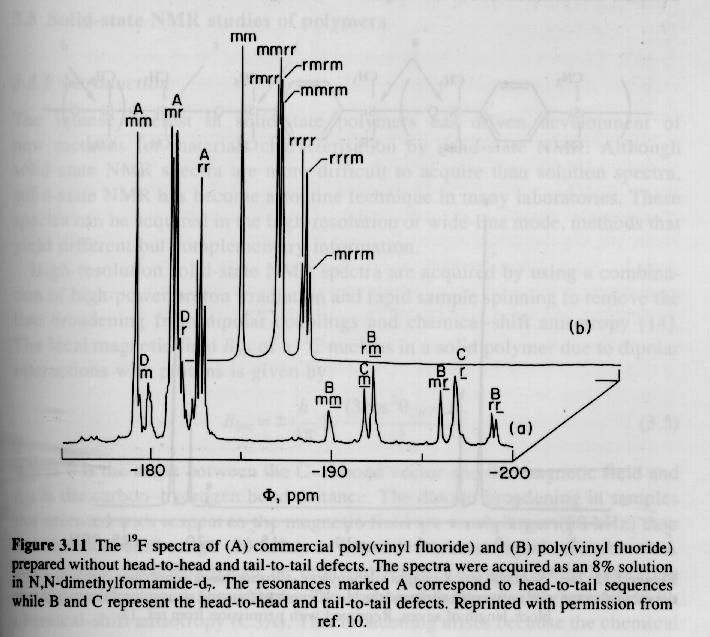
Other Nuclei:
A number of plastics and elastomers are based on nuclei other than 13C or 1H. Two examples are given below, 29Si (natural abundance 4.7%) and 19F (natural abundance 100.0%). Silicon 29 NMR is conducted similar to carbon 13 NMR while Fluorine 19 parallels closely proton NMR. The advantage of using these alternative nuclei is that the degree of C substitution on Si can be directly determined and a much higher resolution of fluorinated tacticity can be determined.