PDF File: (Click to Down Load): Chapter7.pdf
The general principles of diffraction are covered in Cullity, "Elements of X-ray Diffraction". If you are unfamiliar with XRD you will need to review or read Cullity Chapters 1-7 and the appendices. Alexander's text referenced above is also useful as an introduction to XRD but is less general and at a slightly more advanced level.
There are a number of differences between x-ray diffraction in polymers and metallurgical (Cullity) or ceramic diffraction.
1) Polymers are not highly absorbing to x-rays. The dominant experiment is a transmission experiment where the x-ray beam passes through the sample. This greatly simplifies analysis of diffraction spectra for polymers but requires somewhat specialized diffractometer from those commonly used for metallurgy (usually a reflection experiment).
2) DOC: Polymers are never 100% crystalline. XRD is a primary technique to determine the degree of crystallinity in polymers.
3) Synthetic polymers almost never occur as single crystals. The diffraction pattern from polymers is almost always either a "powder" pattern (polycrystalline) or a fiber pattern (oriented polycrystalline). (Electron diffraction in a TEM is an exception to this rule in some cases.)
4) Microstructure: Crystallite size in polymers is usually on the nano-scale in the thickness direction. The size of crystallites can be determined using variants of the Scherrer equation.
5) Orientation: Polymers, due to their long chain structure, are highly susceptible to orientation. XRD is a primary tool for the determination of crystalline orientation through the Hermans orientation function.
6) Polymer crystals display a relatively large number of defects in some cases. This leads to diffraction peak broadening (see Campbell and White or Alexander for details).
7) Polymer crystallites are very small with a large surface to volume ratio which enhances the contribution of interfacial disorganization on the diffraction pattern.
8) SAXS: Due to the nano-scale size of polymer crystallites, small-angle scattering is intense in semi-crystalline polymers and a separate field of analysis based on diffraction at angles below 6deg. has developed (see Alexander and Chapter 8 of these notes for details).
Introduction:
Diffraction or scattering is a separate category of analytic techniques using electromagnetic radiation where the interference of radiation arising from structural features is observed. The interference pattern is the Fourier transform of the pair wise correlation function. The pair wise correlation function can be constructed in a though experiment where a multiphase material is statistically described by a line throwing experiment. If lines of length "r" are thrown in to a 2 phase material there is a probability that both ends of the lines fall in the dilute phase. This probability in 3-d space changes with the size of the line, "r", and a plot of this probability as a function of "r" is a plot of the pair wise distribution function. For a crystal the two phases are atoms and voids and peaks in the pair wise correlation function occur at multiples of the lattice spacing. Interference which results from correlations of different domains or atoms is usually associated with the "Structure Factor" or "Interference Factor", S2(2[theta]). Interference can also occur if the individual domains are prefect structures such as spheres. For a sphere, there is a sharp decay in the pair wise correlation function near the diameter of the sphere and this sharp decay results in a peak in the Fourier transform of the correlation function. For a metal crystal this corresponds to the atomic form factor, f2(2[theta]). For larger scale domains interference associated with the form of the scattering units is generally termed the "form factor", F2(2[theta]).
The scattered intensity as a function of angle is then the product of two terms, the form factor (f2(2[theta]) or F2(2[theta])) and the structure factor (S2 (2[theta])):
I(2[theta]) = Constant F2(2[theta]) S2 (2[theta])
For XRD the form factor is usually obtained from tabulated values and the major interest is in the Structure factor. For small angle scattering dilute conditions are usually of interest making the structure factor go to a constant value of 1 and the form factor for complex structures are investigated.
Thus, the basic principles of scattering and diffraction are the same, while the implementation of these principles are quite different.
Bragg's Law:
Cullity and Alexander derive Bragg's Law using the mirror analogy (specular analogy). It can also be derived from interference laws or using "inverse space" (see appendix in Cullity). The features of Bragg's Law is that structural size is inversely proportional to a reduced scattering angle, so high angle relates to smaller structure and low angle relates to large structure. Small-angle scattering measures colloidal to nano-scale sizes. There is no large scale limit to diffraction. The small scale limit (i.e. the smallest measurable size) is [lambda]/2 as is inherent in Bragg's Law:
d = [lambda]/2 (1/sin[theta])
[theta] is half of the scattering angle measured from the incident beam. The 1/sin[theta] term in Bragg's law acts as an amplification factor. The minimum value of which is 1 for 2[theta] = 180deg. (direct back scattering). The maximum value of the amplification factor is [infinity] so that theoretically no size limit exists with a given radiation of wave length [lambda]. In reality the diffraction geometry and coherence length of the radiation leads to a large scale limit on the micron scale.
Typically diffracted intensity if plotted as a function of 2[theta]. Since the d-spacing is of interest one might wonder why diffraction data isn't plotted as a function of sin[theta] or 1/sin[theta]. This is in fact done with the use of the "scattering vector" q or s. q = 4[pi]/[lambda] sin([theta]) = 2[pi]/d and s = 2/[lambda] sin([theta]) = 1/d. The appendix of Cullity gives a good description of diffraction in "q" or "s" reciprocal space.
The Fourier transform of the real space vector, "r", used to determine the pair wise correlation function is the scattering vector "q".
Review of Crystalline Polymer Morphology:
"Molecular" scale Crystalline Structure:
Consider that we can form an all-trans oligmeric polyethylene sample an bring it below the crystallization temperature. The molecules will be in the minimum energy state and will be in a planar zigzag form. These molecular sheets, when viewed from end will look like a line just as viewing a rigid strip from the end will appear as a line.
Crystal systems are described by lattice parameters (for review see Cullity X-ray Diffraction for instance). A unit cell consists of three size parameters, a,b,c and three angles a, [beta], [gamma]. Cells are categorized into 14 Bravis Lattices which can be categorized by symmetry for instance. All unit cells fall into one of the Bravis Lattices. Typically, simple molecules and atoms form highly symmetric unit cells such as simple cubic (a=b=c, a=[beta]=[gamma]=90deg.) or variants such as Face Center Cubic or Body Centered Cubic. The highest density crystal is formed equivalently by FCC and Hexagonal Closest Packed (HCP) crystal structures. These are the crystal structures chosen by extremely simple systems such as colloidal crystals. Also, Proteins will usually crystallize into one of these closest packed forms. This is because the collapsed protein structure (the whole protein) crystallizes as a unit cell lattice site. In some cases it is possible to manipulate protein molecules to crystallize in lamellar crystals but this is extremely difficult.
As the unit cell lattice site becomes more complicated and/or becomes capable of bonding in different ways in different directions the Bravis lattice becomes more complicated, i.e. less symmetric. This is true for oligomeric organic molecules. For example olefins (such as dodecane (n=12) and squalene (n=112)) crystallize into an orthorhombic unit cells which have a, b and c different while a=[beta]=[gamma]=90deg.. The reason a, b and c are different is the different bonding mechanisms in the different directions. This is reflected in vastly different thermal expansion coefficients in the different directions. The orthorhombic structure of olefinic crystals is shown below. Two chains make up the unit cell lattice site (shown in bold). The direction of the planar zigzag (or helix) in a polymer crystal is always the c-axis by convention.
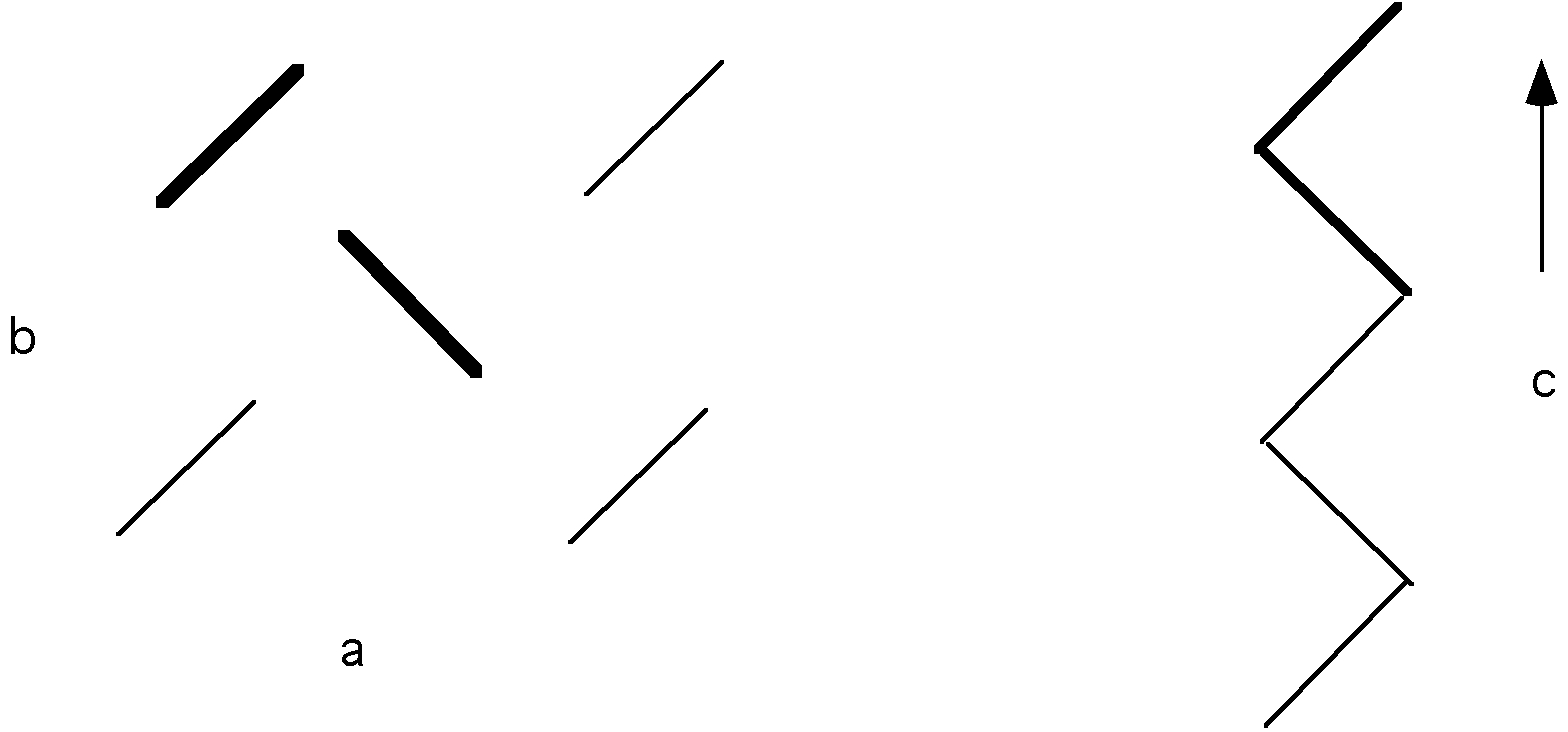
See also, Campbell and White figure 8.4.
Chain Folding:
The planar zigzag of the olefin or PE molecule crystallize as shown above into an orthogonal unit cell. This unit cell can be termed the first or primary level of structure for the olefin crystal. Consider a metal crystal such as the FCC structure of copper. The copper atoms diffuse to the closest packed crystal planes and the crystal grows in 3-dimensions along low-index crystal faces until some kinetic feature interferes with growth. In a pure melt with low thermal quench and careful control over the growth front through removal of the growing crystal from the melt, a single crystal can be formed. Generally, for a metal crystal there is no particular limitation which would lead to asymmetric growth of the crystallite and fairly symmetric crystals result.
This should be compared with the growth of helical structures such as linear oligomeric olefins, figure 4.1 on pp. 143 of Strobl. Here there is a natural limitation of growth in the c-axis direction due to finite chain length. This leads to a strongly preferred c-axis thickness for these oligomers which increases with chain length. In fact, a trace of chain length versus crystallite thickness is a jagged curve due to the differing arrangement of odd and even olefins, but the general progression is linear towards thicker crystals for longer chains until about 100 mer units where the curve plateaus out at a maximum value for a given quench depth. (Quench depth is the difference between the equilibrium melting point for a perfect crystal and the temperature at which the material is crystallized.)
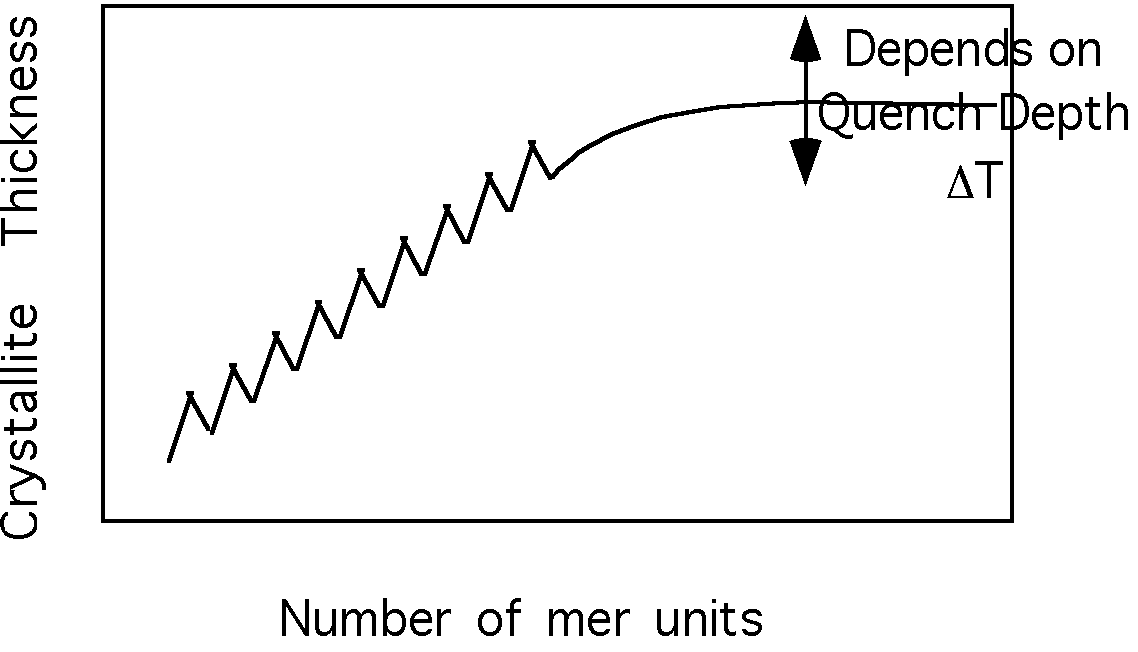
The point in the curve where the crystallite thickness reaches a plateau value in molecular weight is close to the molecular weight where chains begin to entangle with each other in the melt and there is some association between these two phenomena. Also, the fact that this plateau thickness has a strong inverse quench depth dependence suggests that there is some entropic feature to this behavior (pp. 163 eqn. 4.20 where dc is the crystallite thickness and pp. 164 figure 4.18 Strobl).
Considering a random model for chain structure such as shown in figure 2.5 on pp. 21 as well as the rotational isomeric state model for formation of the planar zigzag structure in PE, pp. 15 figure 2.2, it should be clear that entropy favors some bending of the rigid linear structure, and that this is allowed, with some energy penalty associated with gauche conformation of figure 2.2. Put another way, for chains of a certain length (Close to the entanglement molecular weight) there is a high-statistical probability that the chains will bend even below the crystallization temperature where the planar zigzag conformation is preferred for PE. When chains bend there is a local free energy penalty which must be paid and this can be included in a free energy balance in terms of a fold-surface energy if it is considered that these bends are locally confined to the crystallite surface as shown on pp. 161 figure 4.15; and pp. 185 figure 4.34.
There are many different crystalline structures which can be formed under different processing conditions for semi-crystalline polymers (Figures 4.2- 4.7 pp. 145 to 149; figure 4.13 pp. 157; Figure 4.19, pp. 165; figure 4.21 pp. 170). As a class these variable crystalline forms have only two universal characteristics:
1) Unit cell structure as discussed above.
2) Relationship between lamellar thickness and quench depth.
This means that understanding the relationship between quench depth and crystallite thickness is one of only two concrete features for polymer crystals. John Hoffman was the first to describe this relationship although his derivation of a crystallite thickness law borrowed heavily on asymmetric growth models form low molecular weight, particularly ceramic an metallurgical systems. Hoffman's law is given in equation 4.23 on pp. 166:
![]() ,
Hoffman Law
,
Hoffman Law
where n* is the thickness of the equilibrium crystal crystallized at T (which is below the equilibrium melting point for a crystal of infinite thickness, Tf[infinity]), [sigma] is the excess surface free energy associated with folded chains at the lateral surface of platelet crystals, and [Delta]H is the heat of fusion associated with one monomer.
Hoffman's law can be obtained very quickly for a free energy balance following the "Gibbs-Thomson Approach" (Strobl pp. 166) if on considers that the crystals will form asymmetrically due to entropically required chain folds and that the surface energy for the fold surface is much higher than that for the c-axis sides..
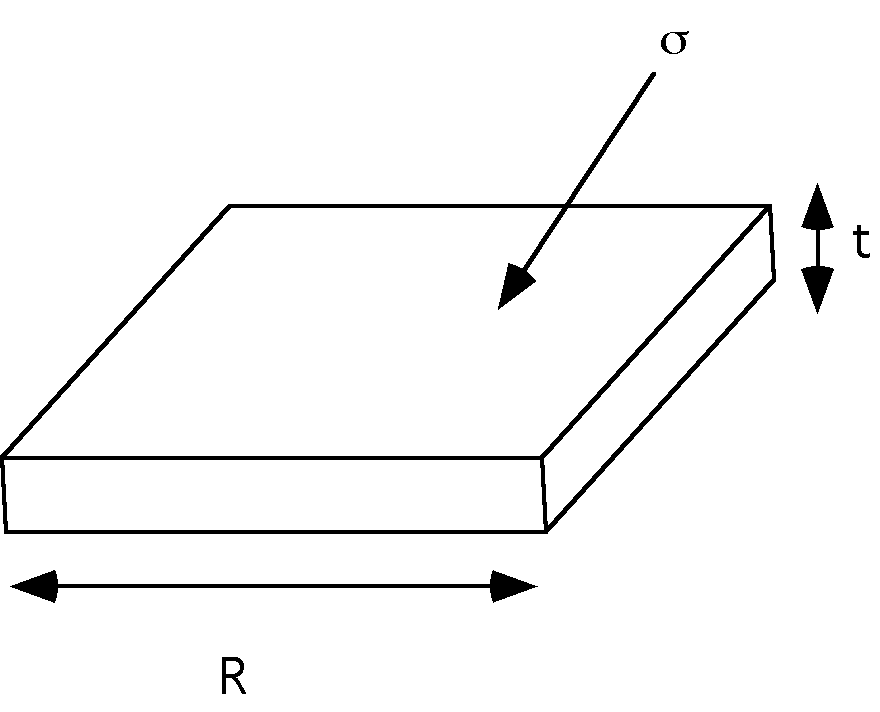
The crystallite crystallized at "T" is in equilibrium with its melt and this equilibrium state is adjusted by adjusting the thickness of the crystallite using the surface energy, that is,
[Delta]GT = 4Rt [sigma]side+ 2R2 [sigma] - R2t [Delta]fT = 0 at T.
That is, At T the crystallite of thickness "t" is in equilibrium with its melt and this equilibrium is determined by the asymmetry of the crystallite, t/R. If [Delta]fT = [Delta]H(T[infinity] - T)/ T[infinity]. is use in this expression,
4t [sigma]side+ 2R [sigma] = R t [Delta]H(T[infinity] - T)/ T[infinity].
Assuming that [sigma]side <<< [sigma], and "t"<<<"R" then,
t = 2 [sigma] T[infinity]./( [Delta]H(T[infinity] - T))
which is the Hoffman law.
The deeper the quench, (T[infinity] - T), the thinner the crystal and for a crystal crystallized at T[infinity], the crystallite is of infinite thickness. (Crystallization does not occur at T[infinity]).
Nature of the Chain Fold Surface:
In addition to determination of T[infinity], the specific nature of the lamellar interface in terms of molecular conformation is of critical importance to the Hoffman analysis. There are several limiting examples, 1) Regular Adjacent Reentry, 2) Switchboard Model (Non-Adjacent Reentry), 3) Irregular Adjacent Reentry (Thickness of interfacial layer is proportional to the temperature).
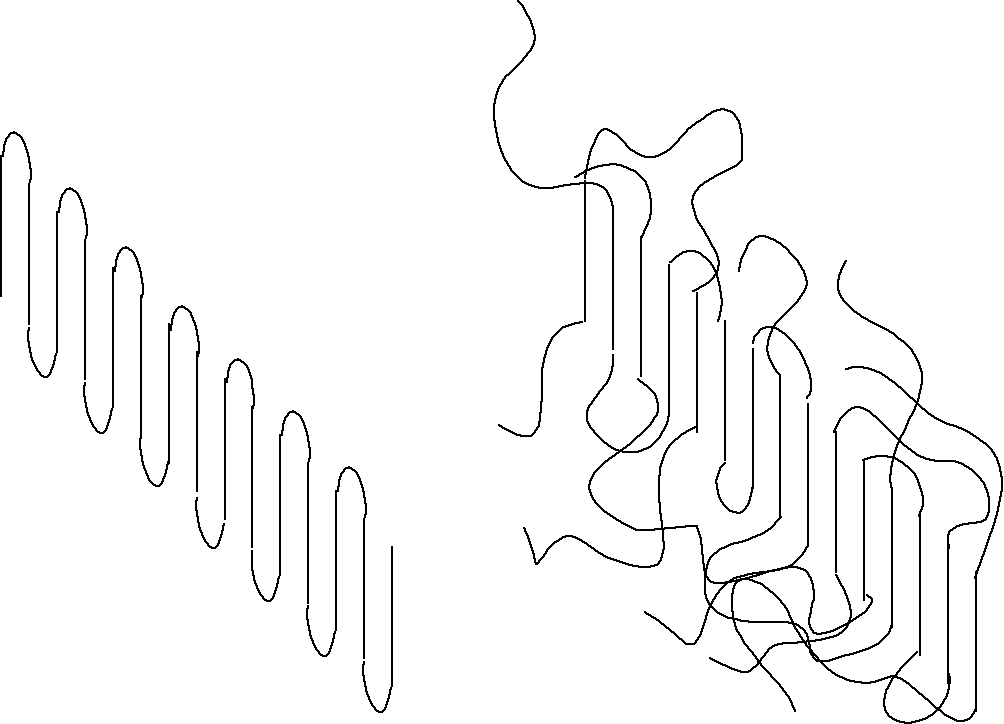

Several distinguishing features of the lamellar interfaces are characteristic of each of these models.
Adjacent Uniform and Thin Fold Surface High Surface Energy
Switchboard Random chains at interface, Broad interface, Low Surface Energy
Irregular Adjacent Temperature Dependent interfacial thickness Intermediate Surface Energy
Interzonal Extremely Broad and diffuse interfaces with non-random interfacial chains
Synoptic Interfacial properties are variable depending on state of entanglement and speed of crystallization.
The Hoffman equation states that the lamellar thickness is proportional to the interfacial energy so we can say that Adjacent reentry favors thicker lamellae since adjacent reentry has the highest interfacial energy and the more random interfacial regions should display thinner lamellae.
Colloidal Scale Structure in Semi-Crystalline Polymers:
Lamellae crystallized in dilute solution by precipitation can form pyramid shaped crystallites which are essentially single lamellar crystals (figure 4.21 for example). Pyramids form due to chain tilt in the lamellae which leads to a strained crystal if growth proceeds in 2 dimensions only. In some cases these lamellae (which have an aspect ratio similar to a sheet of paper) can stack although this is usually a weak feature in solution crystallized polymers.
Lamellae crystallized from a melt show a dramatically different colloidal morphology as shown in figure 4.30 pp. 182, 4.13 on pp. 157, 4.7 on pp. 149, 4.6 on pp. 148, 4.4 and 4.5 on pp. 147 and 4.2 on pp. 145. In these micrographs the lamellae tend to stack into fibrillar structures. The stacking period is usually extremely regular and this period is called the long period of the crystallites.
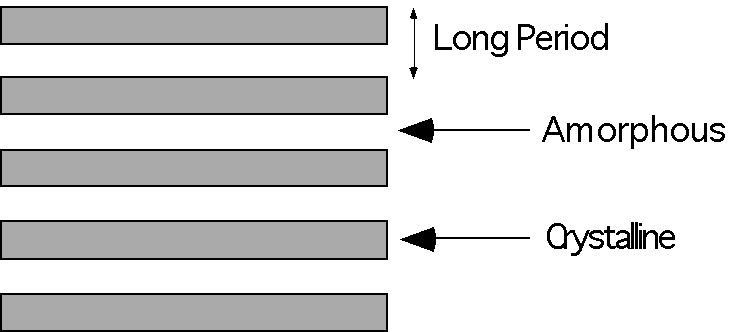
The degree of stacking of lamellae would appear to be a direct function of the density of crystallization, i.e. in lower crystallinity systems stacking is less prominent, and the extent of entanglement of the polymer chains in the melt. You can think of lamellar stacking as resulting from a reeling in of the lamellae as chains which bridge different lamellae further crystallize as well as a consequence of spatial constraints in densely crystallized systems.
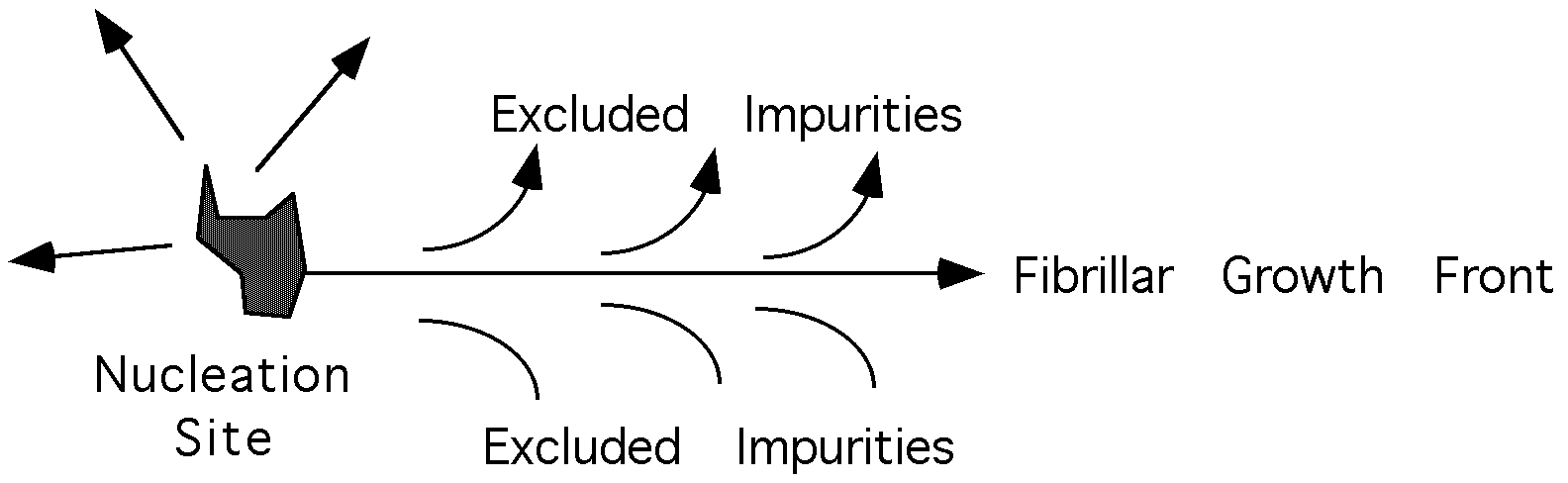
In melt crystallized systems, many lamellar stacks tend to nucleate from a single nucleation site and grow radially out until they impinge on other lamellar stacks growing from other nucleation sites. The lamellar stacks have a dominant direction of growth, that is, they are laterally constrained in extent, so that they form ribbon like fibers. The lateral constraint in melt crystallized polymers is primarily a consequence of exclusion of impurities from the growing crystallites.
Branching of Fibrils: Dendrites versus Spherulites.
Low molecular weight materials such as water can grow in dendrite crystalline habits which in some ways resemble polymer spherulites (collections of fibrillar crystallites which emerge from a nucleation site). One major qualitative difference is that dendritic crystalline habits are very loose structures while spherulitic structures, such as shown in Strobl, fill space in dense branching. At first this difference might seem to be qualitative.
(Incidentally, the growth of dendrites can occur due to similar impurity transport issues as the growth of fibrillar habits in polymers. In some cases a similar mechanism has been proposed where rather than impurity diffusion, the asymmetric growth is caused by thermal transport as heat is built up following the arrows in the diagram on the previous page.)
Non-crystallographic branching leads to the extremely dense fibrillar growth seen in figures 4.4 to 4.7 of Strobl. In the absence of non-crystallographic branching, many of the mechanical properties of semi-crystalline polymers would not be possible. As was mentioned above, non-crystallographic branching may be related to the high asymmetry and the associated high surface area of the chain fold surface which serves as a likely site for nucleation of new lamellae as will be discussed in detail below in the context of Hoffman/Lauritzen theory.
The formation of polymer spherulites requires two essential features as detailed by Keith and Padden in 1964 from a wide range of micrographic studies:
1) Fibrillar growth habits.
2) Low angle, Non-crystallographic branching.
Polymer Spherulites.
Figure 4.2 pp. 145 shows a typical melt crystallize spherulitic structure which forms in most semi-crystalline polymeric systems. The micrographs in figure 4.2 are taken between crossed polars and the characteristic Maltese Cross is observed and described on the following page. The Maltese cross is an indication of radial symmetry to the lamellae in the spherulite, supporting fibrillar growth, low angle branching and nucleation at the center of the spherulite. In some systems, especially blends of non-crystallizable and crystallizable polymers, extremely repetitive banding is observed in spherulites as a strong feature, figure 4.7 pp. 149. Banding is especially prominent in tactic/atactic blends of polyesters and it is in these systems in which it has been most studied. It has been proposed by Keith that banding is related to regular twisting of lamellar bundles in the spherulite (circa 1980). Keith has proposed that this twisting is induced by surface tension in the fold surface caused by chain tilt in the lamellae (circa 1989). Since most spherulites crystallize in an extremely dense manner it has been difficult to support Keith's hypothesis with experimental data. Regular banding has, apparently, no consequences for the mechanical properties of semi-crystalline polymers so has been essentially ignored in recent literature.
XRD of Polymers:
Four main features of XRD are of importance to Polymer Analysis:
1) Indexing of Crystal Structures
2) Microstructure
3) Degree of Crystallinity
4) Orientation
1) Indexing of Crystal Structures: Indexing of crystal structures is similar to the descriptions in Cullity and other metallurgical texts. The main difference is that polymer crystals can not be formed in perfect crystals, so single crystal or Laue patterns are not possible. Also, polymer crystals tend to be of low symmetry, orthorhombic or lower symmetry, due to the asymmetry in bonding of the crystalline lattice, i.e. the c-axis is bonded by covalent bonds and the a and b axis are bonded by van der Waals interactions or hydrogen bonds. Additionally, the unit cell form factor tends to be fairly complicated in polymer crystals. Several unit cells for polymers are shown below:
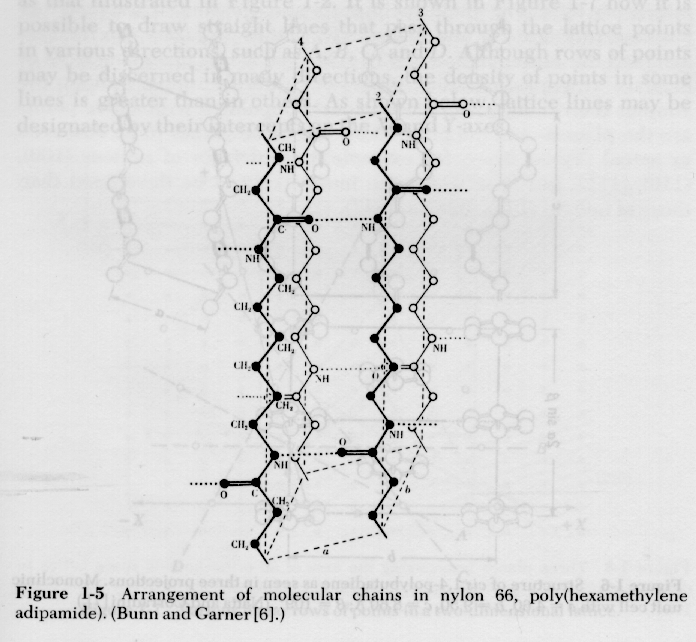
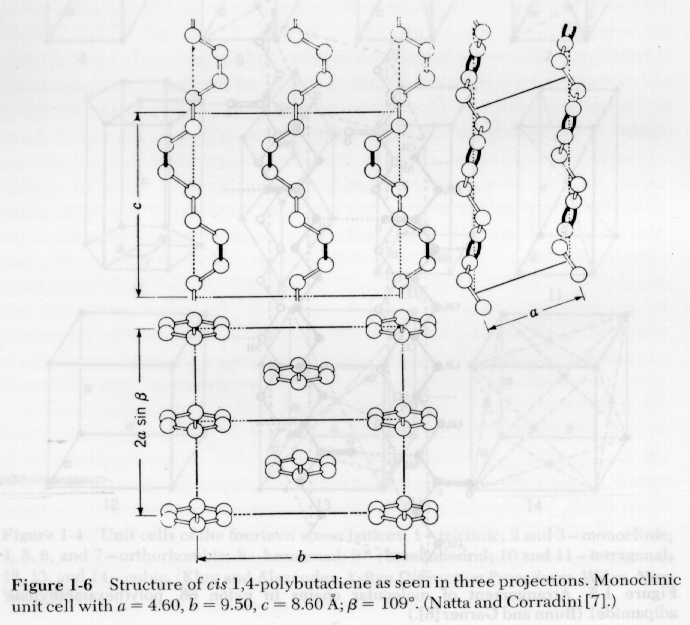
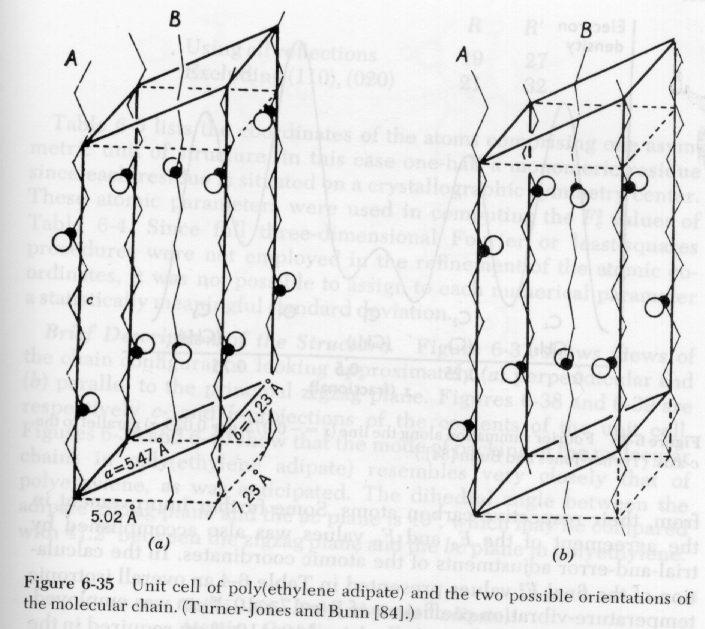
Lattice parameters in polymer crystals are strongly temperature dependent as shown in the following diagram:
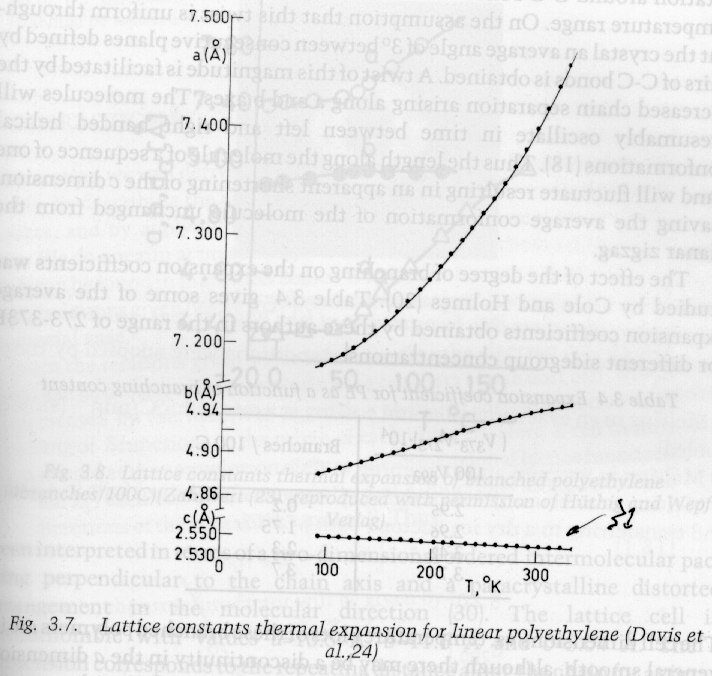
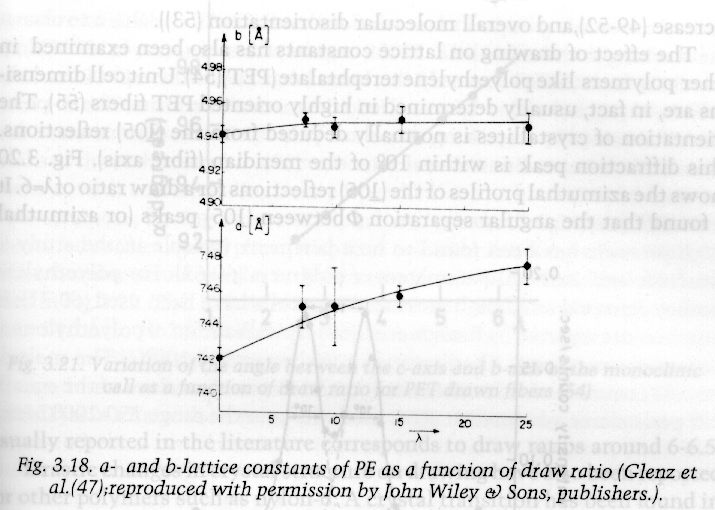
Notice that the c-axis (covalent main chain bonds) is much less dependent on thermal or mechanical strain.
Line widths are broad for polymer diffraction and a substantial amorphous peak is usually present.
2) Microstructure:
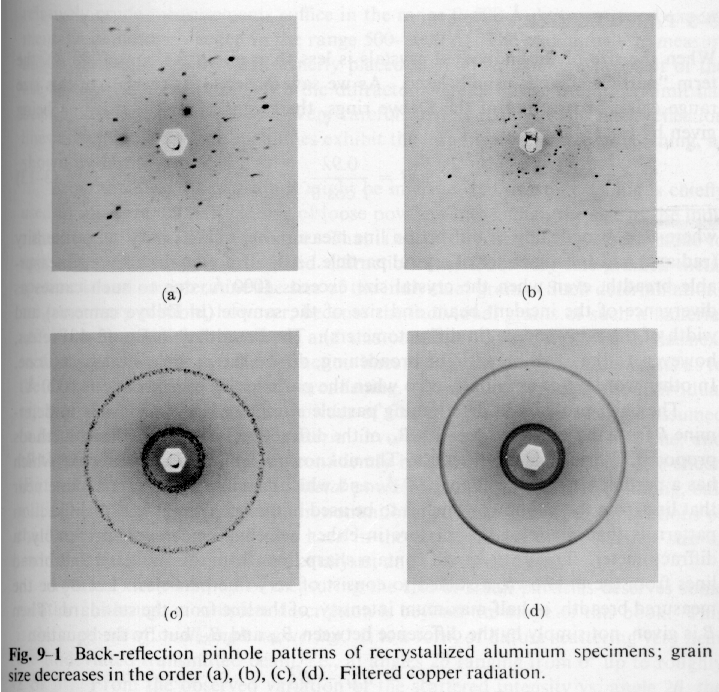
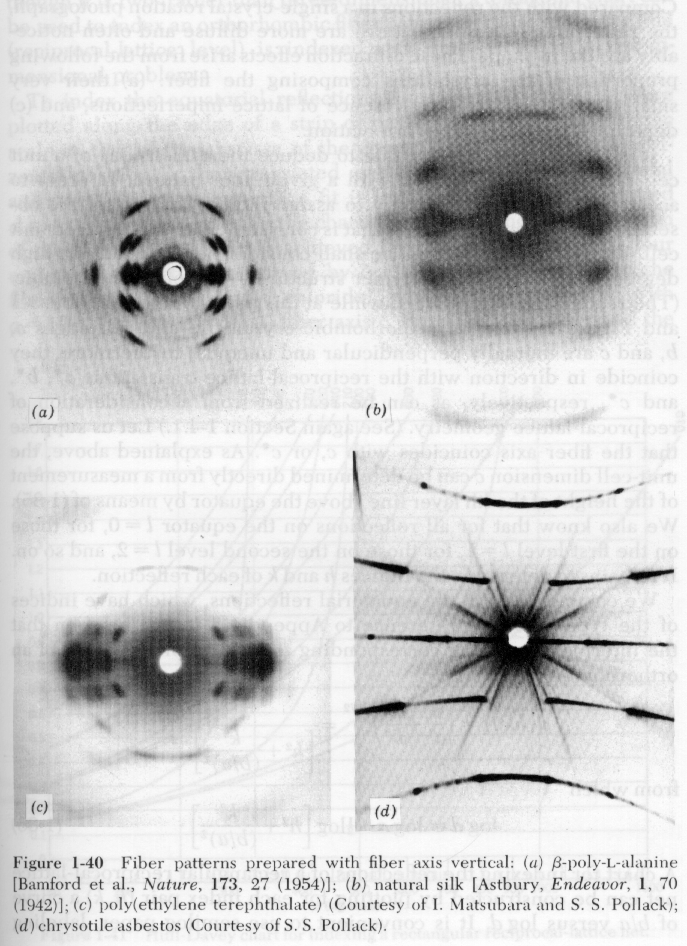
Cullity deals with metallurgical crystals where crystallite sizes are typically larger than a micron. With a monochromatic incident beam the diffraction pattern from a single crystal is a sequence of spots where the Bragg condition is met for certain orientations of crystals (see "a" in figure below). As the crystallite size becomes smaller, more crystallites meet the Bragg condition and the radial orientation of these crystallites cover a broader spectrum of angles ("b" and "c" below), eventually forming Debye-Scherrer powder pattern rings ("c" below). If crystallite sizes approach 0.1 micron (1000Å), the Debye-Scherrer ring begins to broaden ("d" in figure below).
Polymer crystals are on the order of 100Å in thickness. Broadening of the diffraction lines due to small crystallite size becomes a dominant effect and the breadth of the diffraction lines can be used to measure the thickness of lamellar crystals using the Scherrer equation:
![]()
In addition to Scherrer broadening diffraction lines can be broadened in polymers due to defects in the structure. This will not be covered in detail in this course but is described in Campbell and White and in Alexander's text.
3) Degree of Crystallinity:
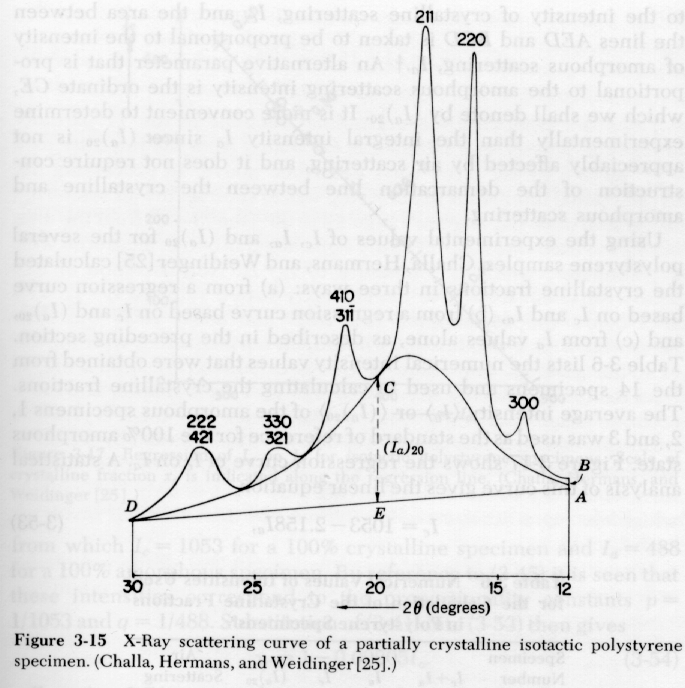
Polymers are never 100% crystalline since the stereochemistry is never perfect, chains contain defects such as branches, and crystallization is highly rate dependent in polymers due to the high viscosity and low transport rates in polymer melts. A primary use of XRD in polymers is determination of the degree of crystallinity. The DOC is determined by integration of a 1-d XRD pattern such as that shown below for polyethylene.
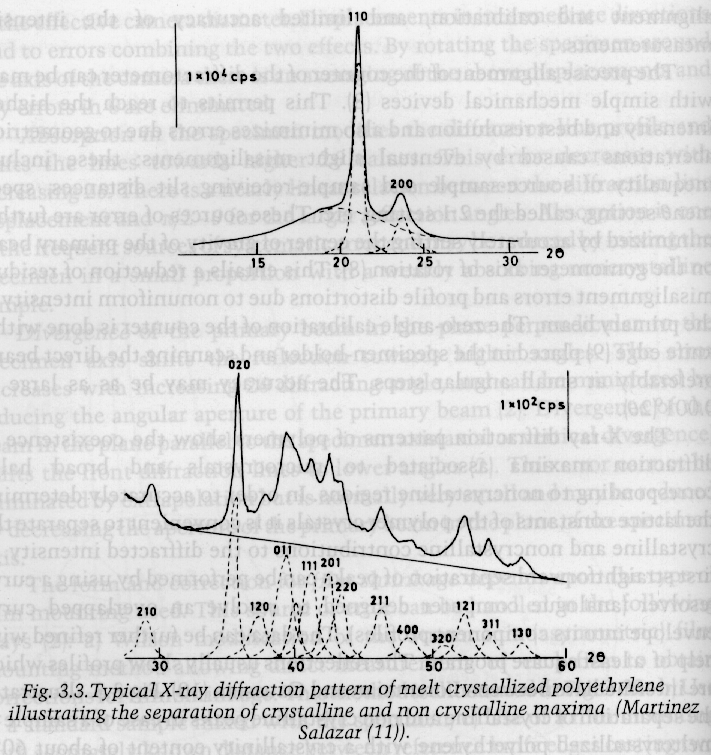
The integrated XRD intensity measures the volume fraction crystallinity, [phi]c. Other techniques such as density gradient columns (see Campbell and White or DSC) measure a mass fraction crystallinity [Psi]c. The two fractions are related by the density ratios, where [rho]c is the crystalline density, [rho] is the bulk sample density and [rho]a is the amorphous density,
![]()
If the density of the sample is known from a density gradient column, the weight fraction degree of crystallinity can be obtained using:
![]()
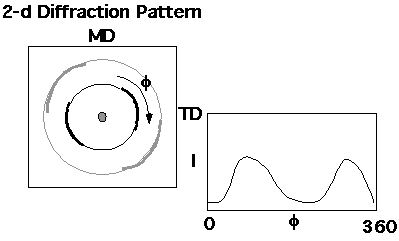
Determination of [phi]c from the XRD pattern under the 2-phase assumption involves separation the diffraction pattern into three parts, 1) Crystalline; 2) Amorphous and 3) Compton Background (Incoherent Scattering). The diffracted intensity if proportional to the amount of each of these contributions. Consider the 2-d diffraction pattern shown just above section 2) above. The 1-d diffraction pattern is a line cut through this pattern as shown below:
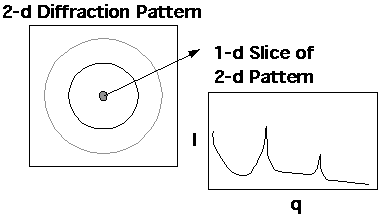
![]()
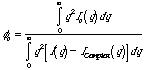
1) Random crystallite orientation (Powder pattern)
2) 3-d crystalline ordering
4) Crystalline peaks can be separated from the amorphous halo
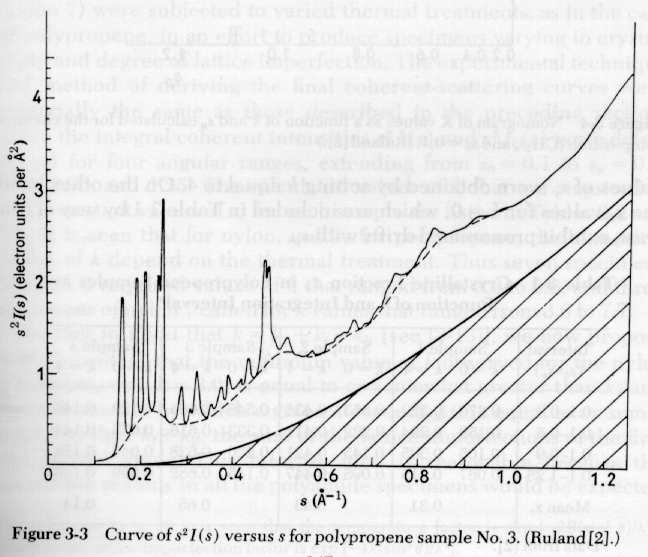
The Ruland equation can be modified for crystalline defects as described in Campbell and White and Alexander. Usually the simple form given above is sufficient. The Ruland method is shown in the figures below where Iq2 is plotted as a function of q (or s).
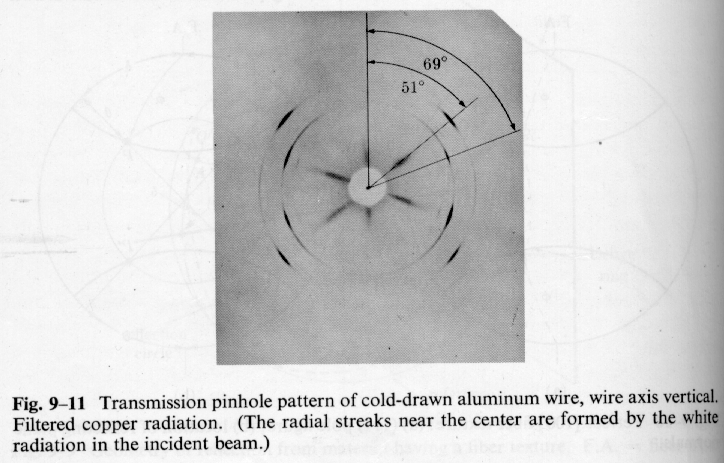
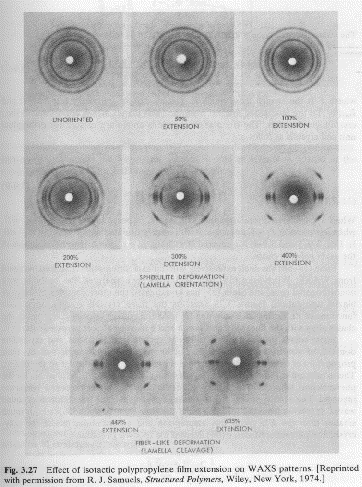
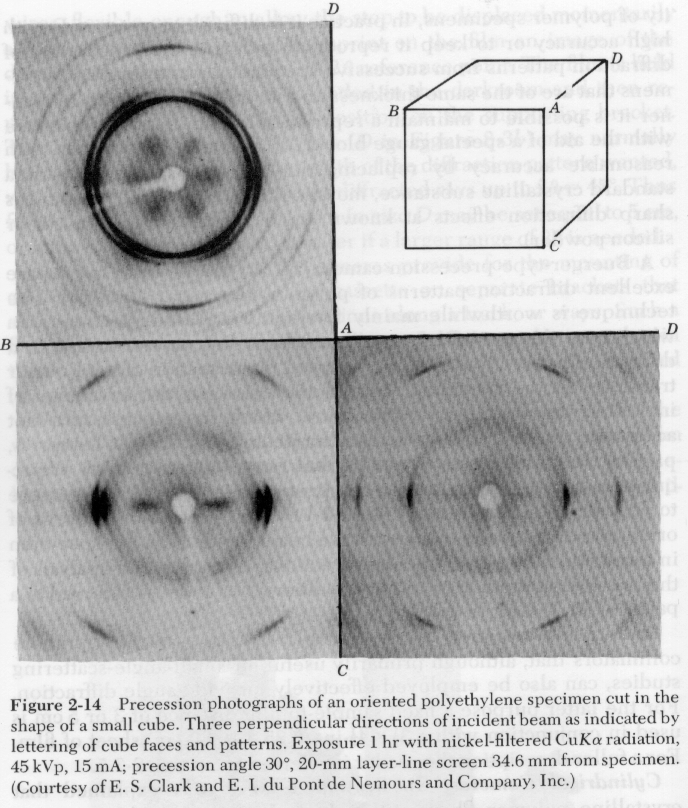
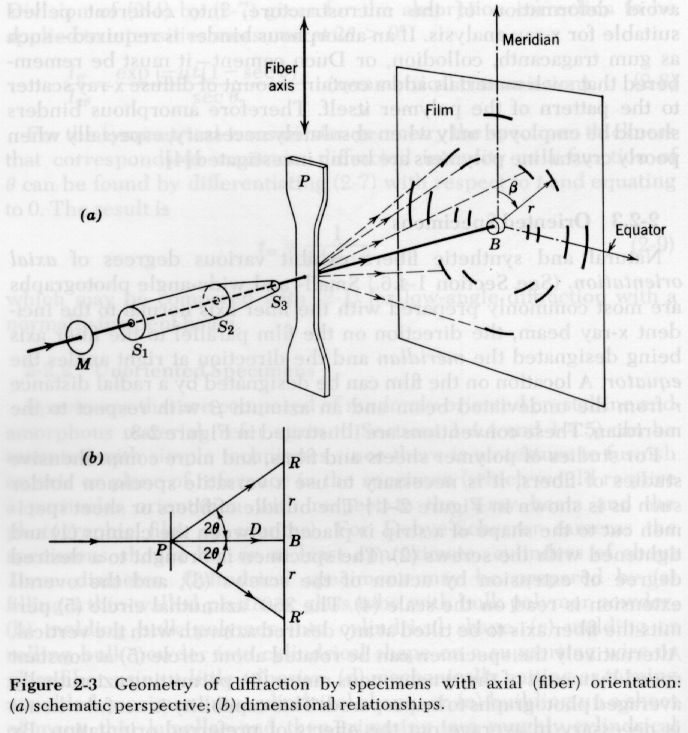
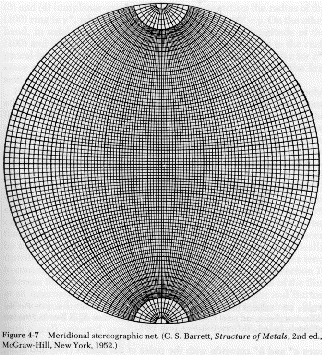
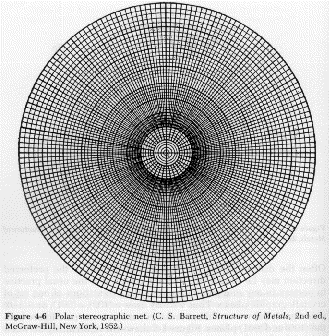
There are a number of techniques for the quantification of orientation from diffraction data. Cullity describes the use of stereographic projections on a Wulff Net (shown below left). The Wulff net is useful if single crystals are studied and it is desired to determine the orientation with respect to the diffraction experiment such as in orientation of semi-conductor samples for cleavage. In most polymer applications it is desired to determine the distributions of orientation for a polycrystalline sample with respect to processing directions such as the direction of extrusion, (machine direction MD), the cross direction (CD) and the sample normal direction (ND). A more useful stereographic projection for these purposes is the polar net or pole figure (shown below right).
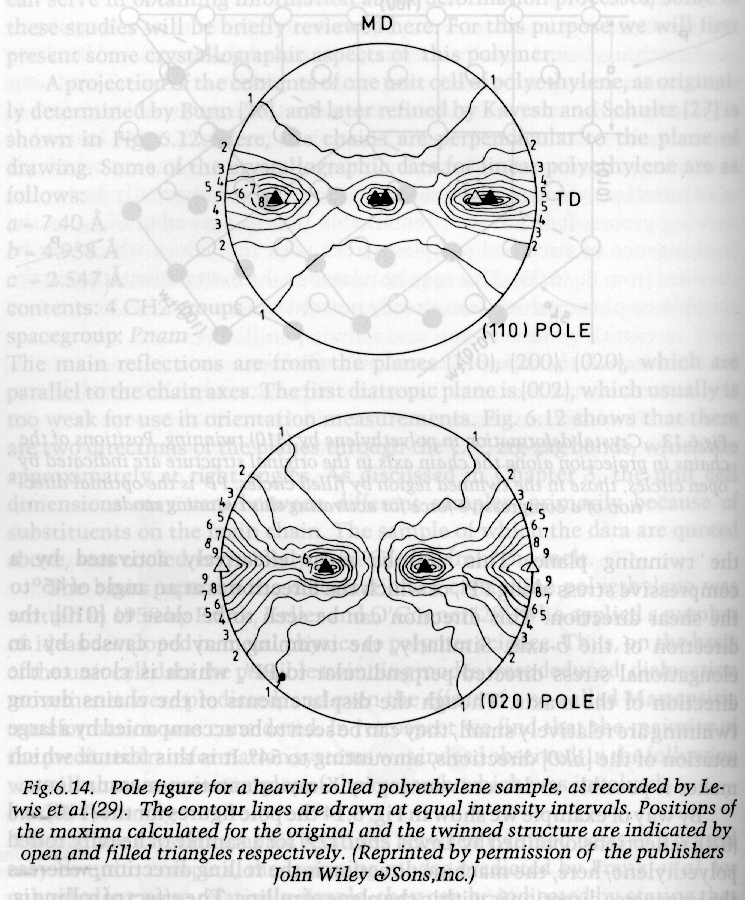
The pole figure is a slice across the equator of the sphere of projection with the MD usually defined at the top of the pole figure and either the CD or ND as the right side. Normals to planes are projected from the south pole to the point of intersection on the sphere of projection and where they cross the equatorial plane a point is plotted on the pole figure. A typical polar figure for a processed polymer is shown in the figure below for the (110) and (020) normals for the polyethylene orthorhombic crystalline structure. Notice that the plane normals appear as a topographical plot since there is a distribution in orientation. The (110) and (020) reflections are the two dominant peaks in the 1-d diffraction pattern for PE shown above (start of section 3).
The following figure shows the type of qualitative analysis of orientation which can be performed using pole figures. Generally, pole figures are constructed by computer software which is part of a diffractometer capable of measurement of pole figures such as the Siemens D-500.
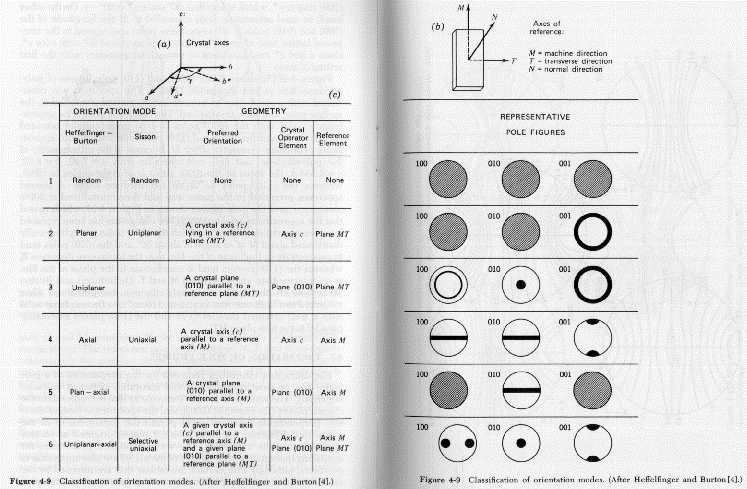
The pole figure can give a qualitative picture of orientation in a polymer sample. Quantitative measures of orientation can be obtained by considering a radial plot of diffraction data.
![]()
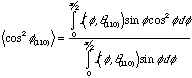
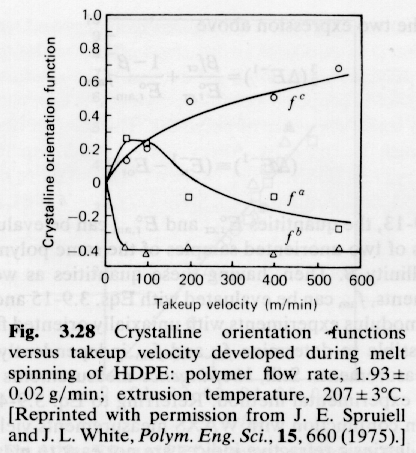
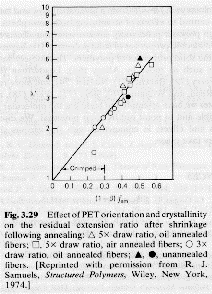
The orientation function is directly related to polymer properties as shown in the example below.