Chapter 1.
Nano-Powders: Organization of the Disordered/NanoCluster Nucleation
Nano-Powders are particulate materials that display dominant structural features on size scales from 1 to 100 nm. In some cases nano-powders are industrially produced in large quantity (tons), for example fumed silica and carbon black, while in other cases they can be extremely precious materials available in only sparing quantities, for example, stabilized metallic quantum dots and atomic clusters. Despite the current level of interest and research in nano-particles, industrial use of nano-powders has been wide spread since the beginning of recorded history. For example the use of nano-sized pigment and carbon colorants was known even with primitive peoples. The current use of nano-powders in industry is extremely broad and there are few commercial products that do not include nano-particles as a component. The use of nano-powders has focused on the special properties that can be achieved with small particles (< 1 micron in size) and these properties include:
Dispersability of an immiscible phase
Uniformity and fine grain size in applications such as photographic and magnetic recording media.
Extremely high surface area per mass
Control over the scattering of light
Enhanced chemical activity of atoms and molecules at an interface
Physical properties associated with small size such as fine abrasives for CMP
Absorptive capabilities of fine particles
Control over microstructure
Transport properties of small domains and pores (Knudsen Transport)
Controlled
electronic states for atoms in nano-sized particles (UV absorption and other
electronic
features)
Many of the properties associated with nano-powders are directly related to the relatively higher energetic state of atoms and molecules at a surface when compared with those in the bulk. This higher energy associated with the surface also drives growth of phases and presents the greatest challenge to the synthesis of nano-sized particles. In many cases the production of nano-powders involves techniques to hinder the natural course of thermodynamics through manipulation of kinetics. Non-equilibrium thermodynamics serves as a basis for the production of kinetically hindered nano-structures. In other cases it is possible to hinder the natural growth of phases through the use of dilution or through protection of surfaces using surface active agents or by coating and encapsulation of nano-particles during growth and coalescence. Viscous entrapment such as immersing nanoparticles in a glassy media is also used in polymers for instance.
Nano-particles lie stratigically between the size scales normally associated with thermodynamics and chemistry, 0.01 to 1 nm, and the size scales associated with macroscopic phenomena. To some extent nano-particles are subject to Brownian thermal motion in an aerosol for instance while they do not entirely display the ability to participate as a species in a chemical reaction. Aggregation is known to occur for nanoparticles that display a high mutual chemical reactivity, but which exist in conditions that hinder chemical growth, such as dilute conditions or low temperature. Under these conditions aggregation can lead to stable, open ramified structures that serve as a useful and efficient form for their industrial use and a simple alternative to more complex dispersion and surface protection schemes. By far, the majority (in volume) of industrially used nano-powders such as fumed silica and titania, precipitated silica, iron oxide and other organic and inorganic pigments display this ramified mass-fractal secondary structure. The growth of these kinetic structures is well understood after close to 30 years of study. In many cases simple growth laws, based on computer simulation and experimental observation, can be used to understand and control nano-aggregate formation. The advantage of nano-aggregates mimics the goals of chemical and physical stabilization of nano-particles, the maintenance and stabilization of high levels of available surface area. Additionally, nano-aggregates provide the chemical and physical properties of nano-particles with the ease of manipulation of micron scale particles.
Since the properties of nano-particles are associated with their size it is generally of interest to control particle size to as narrow a range as possible. For some applications such as silver bromide particles for photographic film and iron oxide particles for magnetic tape, there is a clear industrial need for monodisperse nano-particles. In many cases this need can be relaxed such as for silica filler materials in the plastics industry and in some applications there is an advantage to a controlled dispersion in size.
Nano-particles can be either crystalline or amorphous. Single-grain crystalline nano-particles such as silver bromide and zirconia particles are typically faceted with differences in the surface energy between crystal faces. Multi-grained, such as titania, and amorphous, such as silica, nano-particles are typically spherical in shape and have a homogeneous surface energy. Despite differences in molecular arrangement, many of the growth laws associated with particle and aggregate formation are broad enough to describe both crystalline and amorphous nano-particles. For example, growth laws for crystalline titania nano-particles formed in pyrolytic (flame) synthesis are almost identical to growth laws for amorphous fumed silica. Growth laws of condensing species such as for the carbon sooting reaction, differ dramatically from those for pyrolytic titania synthesis despite their both being multi-grain crystalline nano-particles. The final crystalline state of the nano-particle is often of less importance than the details of kinetics and chemistry associated with the nano-particle formation.
The surface of nano-particles is generally considered to be more disordered than the interior of the particles since the density is generally lower and atoms and molecules are subjected to less constraint at an interface. For this reason, most nano-particles should be considered to be composed of at least two phases. The surface phase is often considered to be similar to an amorphous phase even in faceted crystalline nano-particles.
The Thermodynamics of Nano-Particle Formation (following
Tadao Sugimoto):
Nano-particles are formed in the general sequence of nucleation; growth; Ostwald ripening; Aggregation/Agglomeration/Sintering/Coalescence. All of these processes are dominated by the surface energy of the nano-particle. The interfacial energy is the energy associated with an interface due to differences between the chemical potential of atoms in an interfacial region and atoms in neighboring bulk phases. For nanoparticles the bulk media is generally a solid (the nanoparticle) and a liquid or gas from which the particles grow. The thermodynamics of nano-particle growth mimics in some ways the thermodynamics of polymerization. For instance, the chemical subunit of nanoparticle growth is called a monomer, AgBr for silver bromide particles and SiO2 for silica nanoparticles. If two monomers join they form a dimer, trimer etc. so generically we can speak of an n-mer nanocluster. A dissociated monomer in solution is called a monomeric species, such as Ag+ Br- ions in water. The formation of a free monomer, M, in the media, L, from a solid phase, MS, is described by,
MS + mL -> MLm
where MLm is the solvated monomer.
We can consider the free energy based on a single monomer, M. The free energy of formation for the complex MLm is given by,
![]()
where 0 refers to the pure substance and 0 bar in the first term after the equal sign is the extrapolated molar fraction x = 1. This is the surface energy of a monomer between the media (solution or vapor phase) and solid (or cluster) phase. For a complex at a lower concentration, xMLm, at equilibrium between the media and solid and at dilute conditions where xL is close to 1,
![]()
We can subtract the media chemical potential, ![]() , from the complex in solution,
, from the complex in solution, ![]() , to define the excess chemical potential of the monomer in
solution (in media phase), m, as,
, to define the excess chemical potential of the monomer in
solution (in media phase), m, as,
![]()
Then, at equilibrium,
![]()
So at equilibrium the excess solution chemical potential equals the solid chemical potential at dilute conditions. For such a dilute condition the excess chemical potential in solution can be written,
![]()
and the free energy of a surface monomer is associated with the difference between the excess chemical potential of the cluster at x=>1 and that of the monomer in the solid which is equivalent to the excess chemical potential of the complex at concentration x in equilibrium with the solid,
![]()
where c is the number concentration of monomer and molecular volume of the solvent.
Consider a cluster of size n that is an amorphous solid nano-particle of spherical shape. The surface area of such a cluster, an, is given by,
![]()
where a1 is the surface area of a monomer in the
media (liquid) phase, and v1 is the volume of a monomer in the
solid. The surface energy of such
an n-mer, ![]() , is given by the surface energy of one mer times the number
in the surface,
, is given by the surface energy of one mer times the number
in the surface,
![]()
in the absence of absorbed species. The surface energy per area is given by,
![]()
To form low surface energy particles the number concentration of monomers in the growth media (liquid) phase should be low and this is generally the case.
Modification for Single Crystal Particles:
Single crystals present faces of different crystallographic
index. The main difference between
these faces, in terms of surface growth, is the number of "open
sites" available for growth.
An open site is a position of correct orientation and with sufficient
space and coordination for a monomer to attach to the interface of the
crystal. For a NaCl crystal, for
example, the structure is FCC. An
atom, Na, on a (100) face can bond with one Cl so the number of open sites on
the (100) plane is 1. For the
(110) plane an atom of Na can bond with two Cl's while on the (111) plane the
same Na has 3 bonding possibilities.
The free Na+ ion has 6 open sites since the FCC coordination
is 6 however one of these is taken by an associated Cl ion so the NaCl monomer
has 5 open sites, ns = 5.
(A copper atom attaching to a FCC crystal would have 6 monomer
sites.) The intrinsic surface
energy of a monomer at the surface is the difference in the chemical potential
of a monomer at the surface, ![]() , and of a monomer in the bulk,
, and of a monomer in the bulk, ![]() ,
,
![]()
The surface energy of a free monomer was given above as,
![]()
The surface energy of an open site on the crystal, e, can be obtained by dividing either the surface or the free monomer by the number of sites for the monomer or the specific crystallographic surface of interest,
![]()
We can solve for ![]() ,
,
![]()
where l is the ratio of surface to free open sites,
![]()
The intrinsic specific surface energy of the crystallographic face is given by,
![]()
![]() is called the
surface number density of the surface monomers.
is called the
surface number density of the surface monomers.
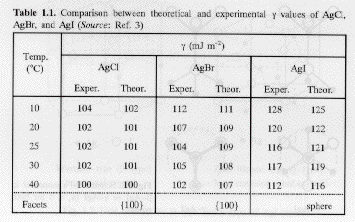
Table 1. Silver Halide surface energy J. Phys. Chem. B 103 3615 (1999).
Gibbs Thompson Equation:
The experimental data in Table 1 was determined using the Gibbs-Thompson Equation for solution growth of nano-crystals. The Gibbs-Thompson equation is commonly used in three forms. The form (sometimes called the Ostwald-Freudlich equation) used to obtain the experimental values in table 1 is,
![]()
x is the mole fraction in the supersaturated solution and x∞ is the mole fraction at equilbrium at temperature T, v1 is the molar volume of a monomer in the nano-particle, and r is the nanoparticle size. By measuring the particle size, r, and the concentration of the solution from which the particle is grown, g can be determined. The Gibbs-Thompson equation is obtained by considering the free energy of formation (nucleation) of an n-mer nano-particle from a supersaturated solution at temperature T,
![]()
where f is the difference in chemical potential for a monomer between the supersaturated solution and the solution in equilibrium with the nanoparticle and A is the surface area of the nanoparticle.
![]()
At equilbrium,
![]()
where v1 is the volume of a monomer and V is the
volume of an n-mer. For a sphere V
= (4/3)pr3 and A = 4pr2 = ![]() so,
so,
![]()
by substitution and solving for x the Gibbs-Thompson equation can be obtained as given above. The equation indicates that smaller particles are more soluble than larger particles. Crystallization at high supersaturations lead to smaller particles.
The Gibbs-Thompson equation indicates that small particles are inherently unstable and indicates that some type of kinetic or physical locking in of unstable particles will be necessary for the formation of extremely small particles from highly supersaturated conditions.
1/r in the Gibbs-Thompson equation is the curvature, k, of the surface, so the second form of the Gibbs-Thompson equation involves the solubility of a surface of curvature k1 and k2, where the subscripts 1 and 2 refer to the principle axies of curvature. The curvature can be positive or negative,
![]()
For surfaces of negative curvature such as the point of contact between two spheres, the Gibbs-Thompson equation indicates rapid solidification, that is, sharp corners with negative curvature will always fill in following the Gibbs-Thompson formula.
The third form of the Gibbs-Thompson equation (also called the Hoffman-Lauritzen equation) deals with crystallization from a melt under supersaturation due to a temperature quench to from nano-scale crystals,
![]()
where B is a geometric factor (generally 2 to 6) and T∞ is the temperature of crystallization for a bulk crystal. This form of the Gibbs-Thompson is derived by considering that at T∞,
![]()
So considering the free energy of a crystal formed at a temperature T under supersaturation (T<T∞) of size r,
![]()
Solving for r yields the Hoffman-Lauritzen equation. This equation indicates that deeper quenches lead to smaller crystals. The Hoffman-Lauritzen function can be rearranged to a simlar form to the other two forms of the Gibbs-Thompson equation,
![]()
Where the last expression assumes a small value for the argument of the exponential, <<1.
The general Gibbs-Thompson rule is that the further from equilbrium crystallization occurs, the smaller are the crystals which form. In all cases crystals susceptible to recrystallization are formed and some form of kinetic entrapment of these unstable crystals is necessary.
Definition of a Critical Nucleus (Following Tadao Sugimoto):
The free energy of nucleation (formation) of an n-mer nano-particle from a supersaturated solution at temperature T,
![]()
where
![]()
m0 is the
chemical potential of a monomer in the bulk (n = ∞) and ![]() is the standard
chemical potential of an n-mer extrapolated to concentration xn = 1
in solution. The supersaturation
energy parameter f, the difference
between chemical potentials of a monomer in the supersaturated liquid and at
equilibrium with the bulk solid, is given by,
is the standard
chemical potential of an n-mer extrapolated to concentration xn = 1
in solution. The supersaturation
energy parameter f, the difference
between chemical potentials of a monomer in the supersaturated liquid and at
equilibrium with the bulk solid, is given by,
![]()
where S is the supersaturation ratio, S = x/x∞. As in the Gibbs-Thompson discussion,
![]()
Then DGn can be written,
![]()
which can be minimized in n to yield a critical nucleus size, n*,
![]()
n* depends on the cube of the ratio of the effective surface monomer energy to the bulk energy difference between the monomer in the supersaturated solution and in the bulk. Lower surface energy or higher bulk difference in energy leads to smaller critical nuclei. This indicates that deeper saturations, bigger f's, have smaller nuclei consistent with the Gibbs-Thompson indication. The maximum free energy is given by,
![]()
Nuclei below n* are unstable and revert to solution while nuclei larger than n* grow since adding monomers lowers the free energy of the cluster. DG* is the activation energy for nucleation.
The growing nuclei of size larger than n* will be initially disperse in size and the number concentration of nuclei at a size n>n*, cne, in equilibrium with the supersaturated solution is given by,
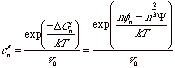
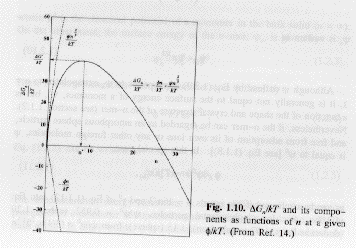
From T. Sugimoto, in "Koroido Kagaku (Colloid Science)" (Chem. Soc. Jpn., Ed.), Vol. 1, pp. 135-152. Tokyo Kagaku Dojin, Tokyo, 1995.
So, n* clusters have the minimum number in the distribution. The larger number concentrations of larger clusters predicted by this thermodynamic description can only be overcome through kinetic limitations. Manipulation of the surface energy by adsorbing additives can lower n* as can driving the system in to a deeper supersaturation state by lowering the temperature.
Kinetics of Particle Formation (Nucleation) from Monomers
(following Tadao Sugimoto):
For an n-mer under equilibrium between deposition of monomers and dissolution of monomers the kinetic equilbrium can be written in terms of two rate constants, kd0 for deposition and kn0 for dissolution,
![]()
where ai is the surface area of an i-mer and c(n) is the number concentration of monomers in equilibrium with an n-mer. (The reaction rate, J, for the two processes is the above terms times the concentration of n-mers, cn.) On the right hand side, kd0 is the rate of incorporation of monomers into the interfacial layer and an-1c(n) is related to the collision frequency. an is related to the surface area of a monomer, "a", by,
![]()
so,
![]()
For the bulk solid n=>∞, so,
![]()
k∞0 and c(∞) are constants (release rate and concentration in equilbrium with bulk solid at T) associated with the bulk solid, so kd0 is independent of n. As noted above, kd0 is the rate constant for the incorporation of monomers into the surface layer of the solid. From the Gibbs-Thompson equation (Ostwald-Freudlich Equation),
![]()
Through substitution and solving for the release rate of monomers from a n-mer, kn0, we have,
![]()
which describes the functionality of the release rate on n, T and the surface energy per monomer, y. kn0 depends strongly on n mostly through the exponential term with a larger effect at small n and dramatically increases with surface energy and decreases with temperature. Small particles grow faster.
This analysis can be generalized to consider several different monomeric species such as various complexes of monomers. A simple analysis can be developed based on an average rate of incorporation of all monomer species, kd,
![]()
C∞, the total number concentration of monomeric species in equilibrium with the bulk solid, and k∞, the overall dissolution rate for all species from the bulk solid,
![]()
A parallel equation for the overall desolution rate for an n-mer per unit area, kn, is given by,
![]()
where,
![]()
For an (n-1)-mer growing to an n-mer and the reverse dissolution process, the forward growth rate, Jn-1,n, is given by,
![]()
where C is the total monomer species supersaturated concentration and cn-1 is the concentration of (n-1)-mers. For the reverse reaction, Jn,n-1,
![]()
Consider monomer-cluster growth where 1-mers grow to 2-mers grow to 3-mers and so forth. The forward rate of growth of the n-mer is given by,
![]()
Generally J is a function of n since an and kn depend on n. If we consider the case where J = 0, that is the system is under quasi-steady state conditions for the sequential reaction, then,
![]()
which yields an expression for J ≠ 0,
![]()
If J were independent of n, the system would be a quasi-steady state system. For small differences in n, J might be expected to behave as such a quasi-steady state system. For this condition and considering a differential change in n, J can be written,
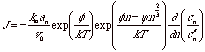
This expression can be simplified (after significant substitution and manipulation) to,
![]()
Using ![]() ,
,
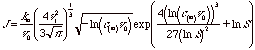
so,
![]()
so,
![]()
for a small range of C. Then a plot of logJ versus logC could yield n* from the slope. From n*, DG*=fn*/2 and y = 3fn*1/3/2 can be calculated.
The number concentration of n-mers can also be determined using this kinetic model under the quasi-steady state assumption. Only the result of this calculation is given here, [T. Sagimoto, Monodisperse Particles, 2001]
![]()
Where
![]()
where cne is the equilibrium number concentration given by the Boltzmann expression previously, and x is the concentration of monomers in equilibrium with an n-mer,
![]()
Then, the number concentration under the quasi-steady state assumption and using a kinetic model, cn, differs drastically from that for equilibrium clusters as shown in the plot of the Boltzmann expression and the expression given above for cn,
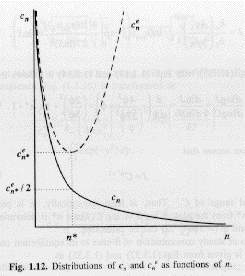
Kinetics can be used to effectively produce relatively narrow number distributions of nano-particles.
In order to achieve quasi-steady state kinetics depletion of monomers from the media must not occur. In the absence of a steady supply of monomers, non-steady state nucleation occurs and Ostwald ripening of clusters. For narrow distributions of small clusters the synthetic scheme should involve some type of reservoir for monomers or must be steadily supplied to the nucleating clusters during the particle formation phase of synthesis. This is achieved in an aerosol reactor, for instance, by supplying small liquid droplets that evaporate during particle growth to form a vapor from which particles form. In this case the entire nucleation and growth step is achieved in seconds. For a solution reactor this has been implemented by Berry [C. R. Berry, J. Opt. Soc. Am. 52, 888 (1962)] in a "double jet" apparatus that provides a continuous supply of monomers in silver halide synthesis. For emulsion based systems, emulsion micelles can supply a steady supply of monomers.