Particle Transport in a Low Density Media:
Nanoparticles are often produced in a gas, plasma or vapor phase. For example, one of the largest sources of nano-particles is pyrolysis of chemical vapors such as SiCl4 or TiCl4. Transport of nanoparticles in low density media (e.g. gas) can differ dramatically from transport in high density media such as liquids depending on the ratio of the mean free path of the media (gas) to the diameter of the particle. This ratio defines the Knudsen number for momentum transfer, Kn,
![]()
where lg is the mean free path of the gas molecules (on the order of 100nm at STP), nm = p/kT, is the molecular density of the gas and s is the molecular diameter. The mean free path is proportional to the gas density so at low pressures the mean free path can be on the order of centimeters (0.01 Torr corresponds to about 1 cm mean free path).
The free-molecular range for particle motion occurs when Kn is larger than 1. In this range the particles are small compared to the mean-free path of the gas and interaction between the particle and the gas can be calculated from molecular collision probabilities.
The continuum range occurs when Kn is smaller than 1. In this regime a zero velocity boundary condition exists since gas atoms leaving the surface of the particle interact strongly with those imparting on the particle. The presence of the zero velocity layer dramatically increases the drag for transport of the particle relative to the gas. The transition between free-molecular and continuum ranges is continuous so intermediate behavior is possible when Kn ≈ 1.
All transport properties display a parallel Knudsen number. For thermal transport, Knudsen thermal range leads to super-insulating porous materials whose thermal conductivity drops exponentially with decreasing pressure. Similar effects are seen for electrical and magnetic properties. Often these Knudsen effects are the source of unique properties for nano-powders. The best understood is momentum transfer.
All suspended particles are subject to diffusive motion and follow Fick's laws,
![]()
For a particle size distribution, nd(dp, r, t)d(dp), the distribution can be inserted on both sides of Fick's second law and divide by d(dp) to describe the evolution of the particle size distribution due to diffusion. D is a function of dp, so the evolution of the PSD depends on the description of D(dp).
Diffusion follows Brownian statistics and the mean square displacement is given by,
![]()
where t is the time of diffusion. This result can be used to calculate the mean square particle velocity, <u2>, through a force balance in one dimension for a Brownian particle,
![]()
where m is the mass of the particle, u is the velocity, t is the time, f is the friction coefficient and F(t) is a fluctuating force due to thermal motion of the surrounding fluid or gas. This equation relies on the definition of the friction coefficient, f. For spherical particles with Kn<<1 (large particles in the continuum range), f is given by Stokes Law,
![]() Continuum
Range
Continuum
Range
where m0 is the matrix viscosity. For the free molecular range f will be calculated below.
F(t) is associated with the surrounding gas or liquid and is independent of u. <F(t)> = 0 since it is a random force. F(t) fluctuates more rapidly than the particle motion.
For a single particle diffusing from a point x = 0 to a point x we can write,
![]()
where A(t) = F(t)/m and b = f/m. If x is inserted in the differential we have,
![]() or
or ![]()
since dx/dt = u. This second order differential equation can be integrated from t = 0 to t (ux = 0 to ux) to yield,
![]()
Averaging over all particles in the system (all starting at x = 0) knowing that <A(t)x> = 0,
![]()
Since u = dx/dt we have,
![]()
Then,
![]()
and integrating from t = 0 to t,
![]()
b is the friction coeffiecient divided by the particle mass which yields the inverse of a time constant, for times much longer than 1/b we have,
![]()
The velocity of the particles is due to the thermally driven impact of the gas (fluid) molecules. Then the higher the temperature the larger is <u2>. Following the equipartitoining of energy (Einstein), we have,
![]()
For Brownian motion we have <x2> = 2Dt so,
![]() or
or ![]() Brownian
Particles (Free or Continuum)
Brownian
Particles (Free or Continuum)
This function (Einstein function) for the friction coefficient does not give a relationship with dp. Such an expression can be obtained from the kinetic theory of gasses by definition of an accommodation coefficient, a, that gives the fraction of gas molecules that leave a particle surface after equilibrating with the surface, i.e. after imparting their excess thermal energy to the particle (typically a ~0.9 for nanoparticles but should be determined experimentally for a given case). Put another way, the particles that elastically collide with the particle is (1-a). The function for f(dp) using a is (Epstein, Phys. Rev. 23 710, 1924),
![]() Free
Molecular Range
Free
Molecular Range
which can be compared with Stoke's Law, f = 3pm0dp. For a 20Å particle the friction coefficent for the free molecular range is about 1% of that for the continuum range. This is mostly due to the dependence on dp2.
When the Knudsen number is close to 1 a modified Stokes Equation can be used to calculate the friction factor that is due to Millikan (in oil drop measurements),
![]()
A1, A2 and A3 are constants. For a = 0.84 Davies found A1 = 1.257, A2 = 0.400 and A3 = 0.55 (Davies, CN, Proc. Phys. Soc. 57 259 1945).
Nanoparticles can deviate significantly from the spherical shape that is the basis for all of the transport equations given above. We can consider three typical deviations, prolate ellipsoids, oblate ellipsoids and mass fractal aggregates following Friedlander.
D for Ellipsoids:
The diffusion coefficient for an ellipsoid is less than that of a sphere. In order to calculate the diffusion coefficient for an ellipsoidal particle we calculate the diffusion coefficient for a spherical particle with the same volume as the ellipsoidal particle, D0. We also determine the ellipsoidal aspect ratio z = b/a where a and b are the major axes of the ellipsiod (the equivalent sphere radius is given by a0 = a z2/3). Perrin (Perrin F, J. Phys. Radium, Series 7, 7 1 1936) gave the following expressions:
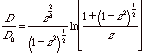 Prolate
(Cigar) z > 1
Prolate
(Cigar) z > 1
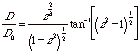 Oblate
(Pancake) z < 1
Oblate
(Pancake) z < 1
For 10>z>0.1, D > 0.6 D0 so the effect is less than critical. Perrin's forumlas are for randomly arranged ellipsiods. If the ellipsiods orient in a shear field the effect is much greater.
D for Mass Fractal
Aggregates:
Individual nanoparticles after formation exhibit a high surface charge per volume, are capable of displaying Van der Waals interactions (attractive) and retain some degree of chemical reactivity due to their disordered surface and high surface energy. Once even a weak bond such as a Van der Waals interaction occurs, the Gibbs Thompson equation (second form) drives rapid reinforcement of the contact point. This occurs for particles smaller than a micron. For these reasons fairly strong bonds can be made between nanoparticles, so clusters of nanoparticles can grow into aggregates. Nanoparticles are subject to diffusive drag, and the time constant associated with bonding between particles decreases rapidly with particle size so diffusion limited growth of these aggregates is common, leading to open, ramified, mass-fractal nano-aggregates of n-mer clusters. For such an aggregate, between the cluster (primary particle) size and the aggregate size, mass scales with size to a mass-fractal dimension that is often close to 1.8 for diffusion limited aggregation. Such an aggregate is similar to a branched polymer chain except that thermal randomization of the structure (entropic control of structure) does not occur. Generally these are rigid, Hookean objects (Friedlander, 2000).
If the time constant for bonding is longer or the bonds are weak, higher dimension, reaction controlled aggregates appear. The mass fractal dimension of such aggregates is typically 2.5. In carbon black synthesis 3-d, solid nano-aggregates sometimes form. Also, rigid aggregates on the order of 0.1 micron in diameter or larger, can retain high surface charge to mass ratios and can loosely bond to form agglomerates of aggregates. These differ partly because Gibbs-Thompson reinforcement of contact points is less dominant and because they tend to form at a later stage of the process where the conditions for bonding are less favorable. For such slow reaction conditions, reaction limited growth is common and the larger scale structures tend to be of high mass-fractal dimension. Processing or mixing can shift this picture by breaking up agglomerates down to about 1 micron size and stripping aggregates of their branches down to about 0.1 micron in size. At smaller scales it appears difficult for processing (shear) to modify the rigid aggregates.
It is of interest to understand the transport properties of such rigid aggregates since they are common in the nano-particle field. Nanoaggregates are useful because at low mass-fractal dimension they retain most of the accessible nano-cluster surface area, they are stable to processing due to their close to micron size and rigid structure, and their inherent Hookean elasticity can be advantageous in many applications where mechanical properties and integrity of the material is of importance.
Polymer diffusion in the limit of dilute concentrations is governed by the extent of penetration of the solvent into the polymer chain (see for instance Heimenz, Polymer Chemistry for a simple treatment of polymer solution viscosity and G. Strobl Polymer Physics for a slightly more advanced treatment). If the solvent fully penetrates the chain then the coil is called "free draining" and the Rouse theory is applicable where the friction coefficient is given by the sum of friction coefficients of the monomers for the chain. At the opposite extreme, for a "non-draining" chain the diffusion coefficient for a chain scales drops off much more rapidly with aggregate size. Degennes (Scaling Concepts in Polymer Physics for example) describes this behavior by, D ~ N-1/df. This is a restatement of Stokes law, f = 3pm0dp, and Einstein's Fluctuation Dissipation Theorem, D = kT/f, where d ~ N1/df for a mass-fractal structure. Then the main issue for nano-aggregates in terms of diffusion involves the extent of penetration of the media into the structure. This, in turn, is decided by the Knudsen number for the aggregate/media system. For high Kn relative to the aggregate, the media can easily penetrate the aggregate and diffusion is dominated by Rouse behavior,
D ~ D1/Nagg
= (d1/dagg)df D1 Free Molecular Range
For low Kn, large aggregates and dense media (liquids), the aggregates are non-draining and
D ~ D1/Nagg1/df = (d1/ dagg) D1 Continuum Range
The intermediate regime is difficult to consider and we should consult the polymer literature such as Doi and Edwards' text on polymer dynamics.
Brownian Motion of Nanoparticles:
A consideration of Brownian motion of nanoparticles can begin with the motion of rigid spherical ideal gas molecules. For such gasses the mean free path, lp (≈ 0.1 micron at STP), for the gas molecules can be directly calculated from the kinetic theory of gasses, (Chapman and Cowling Mathematical Theory of Nonuniform Gasses, Cambridge Univ. Press 1952),
lp = 1/(p√2 Mass d2) Kinetic Theory of Gasses
where Mass = P/kT. The kinetic gas has extremely low mass, so the random thermal motion has little momentum driven "persistence of velocity". That is, it is correct to assume that the motion is composed of small, randomly directed steps. For a 0.01 micron particle of silica and using the kinetic theory the mean free path between the gas and particle collisions is on the order of 1 Å. A heavy particle, however, shows a greater degree of correlation in direction from one instant of time to the next due to momentum. This leads to a higher degree of persistence of velocity.
For a Brownian nano-particle the jagged path of a thermally driven atom becomes, on the size scale of gas atoms, analogous to the smooth path of a particle in turbulent flow. If one views the path on a larger scale and measures it with line vectors connecting points on the path, the path retains the Brownian jagged feature of thermally driven gas atoms. The diffusion coefficient is given by,
D = lpa √<u2>
where lpa is the persistence length for the nanoparticle motion and <u2> is the mean square velocity, <u2> = kT/m (equipartition of energy function). The nanoparticle also follows the Stokes-Einstein relationship, D = kT/f, so,
![]()
The persistence length is also defined by the velocity correlation coefficient, R(q), which is the dot product of the unit velocity vector at different times with the unit velocity vector at a fixed time, q = 0,
![]()
Using, ![]() ,
and friction coefficient definitions for continuum (3pmd) to free molecular ranges, the persistence length, or
particle path length, can be calculated as a function of partile size, dp. For dp < 0.1µm lpa
follows a power law of dp-1/2, since f ~ dp2
and m ~ dp3.
For particles larger than about 0.5 µm, dp follows a
power law dp1/2 according to Stokes law in the continuum
range. A minimum path length is
seen at about 0.2µm for particles with a density of 1g/cc of about
7nm. For 1 nm particles the path
length is about 60 nm.
,
and friction coefficient definitions for continuum (3pmd) to free molecular ranges, the persistence length, or
particle path length, can be calculated as a function of partile size, dp. For dp < 0.1µm lpa
follows a power law of dp-1/2, since f ~ dp2
and m ~ dp3.
For particles larger than about 0.5 µm, dp follows a
power law dp1/2 according to Stokes law in the continuum
range. A minimum path length is
seen at about 0.2µm for particles with a density of 1g/cc of about
7nm. For 1 nm particles the path
length is about 60 nm.
Simultaneous Migration and Diffusion of Nanoparticles:
Nanoparticles are often subject to electrical, magnetic, and
thermal fields that drive migration.
Additionally, the gravitational field often leads to segregation of
certain size particles and this is used to separate particles in sedimentation
processes. The drag force, fc, where c is the particle velocity, opposes the force, ![]() ,
associated with the field, f. For a steady state condition, F = fc, and the steady state migration velocity, c, can be calculated. For example, the terminal settling velocity, cs,
is calculated from the gravitational force,
,
associated with the field, f. For a steady state condition, F = fc, and the steady state migration velocity, c, can be calculated. For example, the terminal settling velocity, cs,
is calculated from the gravitational force,
![]()
yielding,
![]()
The rate (or flux) of settling, Jx, is obtained from,
![]()
Using conservation of mass, ![]() ,
,
![]()
Consider the sedimentation of particles through a thin layer of gas onto a plate. The particles which approach within a diameter or so of the plate are bonded immediately by Van der Waals interactions leaving a concentration of 0 at the interface, z = 0. The bulk concentration is nb at z = b. Gravity acts in the negative z direction so,
![]()
where cs is proportional to dp2 as given above for steady state conditions. The flux equation (first order linear differential equation) can be integrated across the gas film from z = 0 to z = b to yield the integrated sedimentation,
![]()
To yield two limits result from this equation, the diffusion
limit where D >> b cs, and the exponential can be expanded,
exp(-x) = 1 - x + x2/2! - … t to yield, ![]() , and the sedimentation limit where b cs >>
D, so J = -csnb.
In the diffusion limit the sedimentation is higher for smaller particles
since D ~ kT/f and f increases with dp. In the sedimentation limit, sedimentation is higher for
larger particles since cs increases with dp. A minimum rate of deposition exists for
an intermediate particle size. For
a 1 mm diffusion layer particles of 0.1µm diameter are the slowest to
sediment. This can be obtained by
either considering the minimum of the function for J or by equating the
sedimentation and diffusion fluxes and solving for dp. Sedimentation, in general, involves
unexpected fractionation of the particle size distribution.
, and the sedimentation limit where b cs >>
D, so J = -csnb.
In the diffusion limit the sedimentation is higher for smaller particles
since D ~ kT/f and f increases with dp. In the sedimentation limit, sedimentation is higher for
larger particles since cs increases with dp. A minimum rate of deposition exists for
an intermediate particle size. For
a 1 mm diffusion layer particles of 0.1µm diameter are the slowest to
sediment. This can be obtained by
either considering the minimum of the function for J or by equating the
sedimentation and diffusion fluxes and solving for dp. Sedimentation, in general, involves
unexpected fractionation of the particle size distribution.
Electrical Migration:
When nanoparticles are produced in an electric field controlled flame, plasma, arc or other conditions that are conducive to particle charging and the presence of an electric field, migration due to the electric field should be considered. The force on a charged particle, i e, in an electric field, E, is given by the charge times the field,
F = ieE
At steady state this force is balanced by drag, similar to the sedimentation analysis, yielding,
ce = ieE/f
The electrical mobility is defined as Z = ce/E = ie/f.
Charging can occur due to ion attachment, static charge buildup, and thermionic charging (heating the particles to a point of electron or ion release). Exposure to ions of a single charge leads to unipolar charging while exposure to mixed ions can lead to bipolar charging of particles. Particle charging can lead to singly charged particles or multiple charge particles. In general the fractionation behavior seen for sedimentation is also observed for charged particles in an electric field. In general analysis of of particle charging and migration is complex.
Thermophoresis:
Small particles present in a temperature gradient are driven away from regions of higher temperature. For Kn>>1, the media atoms impact the particle at a higher rate on the hot side driving the particles towards the cold end of the temperature gradient. The thermophoretic velocity, cT, is independent of particle size and is given by,
![]()
where a is the accommodation coefficient (used previously) and n is the kinematic viscosity.
For Kn ≤ 1, thermally driven motion is also observed although the mechanism must be different from particles where dp<<lp. Equations have been developed that describe thermal motion for these larger particles based on a continuum model with a creep velocity governed by the slip conditions at the surface of the particle. A plot of reduced thermophoretic velocity as a function of Kn is shown below. The plot shows the independence of thermophoretic velocity at high Kn (nanoparticles) and a decay for large particles in the continuum range. Thermophoresis is often the best mechanism to sample complex gas-phase synthesis of nanoparticles. Thermophoretic sampling involves insertion of a cooled sampling surface into the process for a short period of time, typically shot through the process stream. Thermophoresis drives particles to the surface and they are then bound to the surface either by Van der Waals forces or by charge. Thermophoretic deposition can also lead to processing problems since nanoparticles will be driven to cooler processer walls and lead to formation of fouling deposits and cakes.
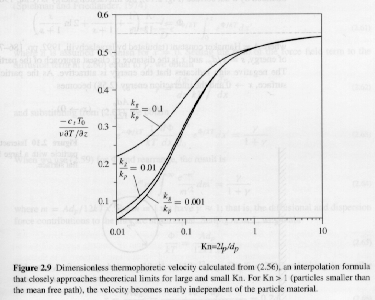
Figure from S. K. Friedlander, Smoke Dust and Haze, 2000.
Van der Waals Forces:
The behavior of nanoparticles with stable chemical species and in the absence of permanent charges are dominated by Van der Waals interactions. These interactions are larger for particles that display conjugation and high polarizability but are strong even for ionically bonded nanoparticles. The Van der Waals force (London force) arises due to the ability of electrons in a nanoparticle or molecule to move randomly in the particle or molecule driven by thermal energy. Van der Waals interactions also rely on the short range nature of dipole/dipole interactions, where the energy of interaction, f, goes as 1/r6, r being the separation distance of two dipoles (from quantum mechanics). The forces also rely on random thermal motion of the nanoparticle or molecule. For two such particles it is possible for temporary dipoles to be arranged with opposite poles aligned at any given instant of time and for these two dipoles to be within a short distance of each other. The result is that the two dipoles will be driven closer to each other by the attraction of opposite poles. At the next instant of time the aligned dipoles could have any charge distribution but it is more likely that they would have a similar aligned arrangement, leading to a net attractive force that is large due to the 1/r6 dependence. Just as likely is to have two particles with misaligned dipoles. The force would drive these dipoles apart leading to a much lower repulsive force in the next instant of time. When these forces are averaged a net attractive force results due to the short range nature of dipole-dipole interactions.
For a spherical particle with Haymaker constant, A, that is near an infinite surface, and with a distance of approach of the particle to the surface being x, we can define a dimensionless parameter, s = x/dp. The attractive interaction energy due to Van der Waals interaction is given by (B. Chu, Molecular Forces: Based on the Baker Lectures of P. J. W. Debye, 1967),
![]()
As x => 0 the interaction energy goes to -Adp/(12x). The interaction energy goes to infinity as a nanoparticle approaches a perfectly smooth surface. A surface is a sink for nanoparticles.